

2024-11-24

近期,动物生态专业委员会委员、中国科学院西北高原生物研究所张同作研究员团队以中国境内奇蹄目(Perissodactyla)和鲸偶蹄目(Cetartiodactyla)的17个常见有蹄类物种为研究对象,通过大规模宏基因组测序和组装、分箱和注释,成功构建了中国典型有蹄类物种的肠道微生物宏基因组图谱,其中共获得131,416个宏基因组组装基因组(MAGs),包含22,998个高质量MAGs和11,175个菌种水平基因组(SGBs),且60%左右的SGBs为首次报道。
本研究不仅进一步阐明了肠道微生物群的功能基因、基因组结构变异和进化模式之间的关系,更深入地揭示了高原有蹄类动物及其肠道微生物群对恶劣高原环境的协同适应机制,为理解肠道微生物功能和遗传演化及其与宿主之间相互作用及共同适应环境提供了新视角。
本研究的宏基因组测序和部分结果分析由上海派森诺生物科技股份有限公司完成。
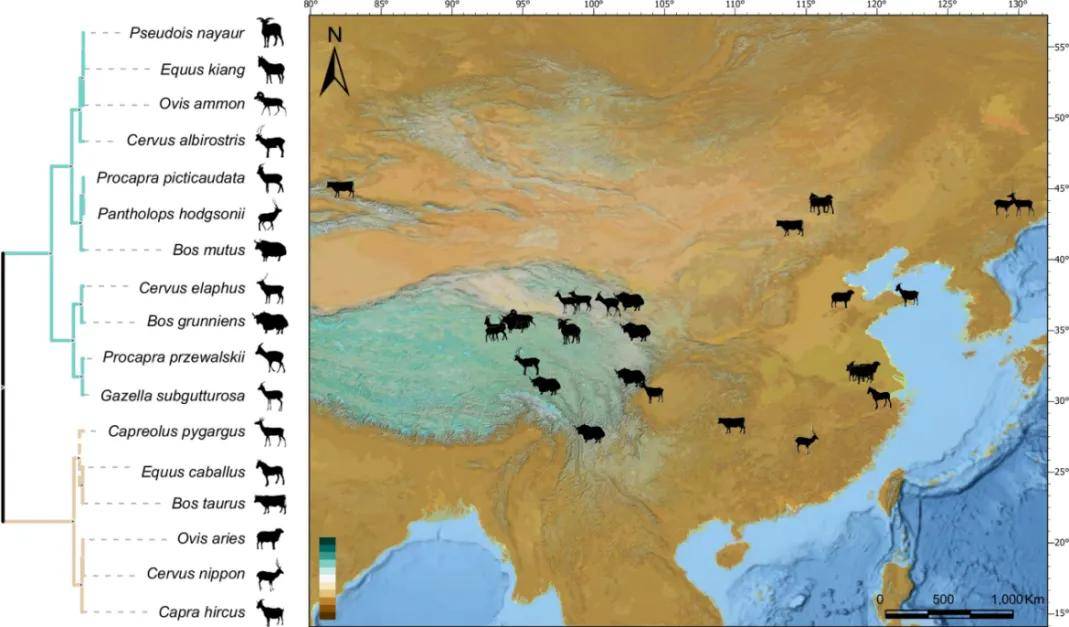
图1 | 本研究中17种常见有蹄类动物的分布情况
研究背景
有蹄类动物在生存和繁殖过程中,主要通过肠道微生物群发酵合成的短链脂肪酸(SCFAs)和挥发性脂肪酸(VFAs)等代谢产物满足能量需求。与平原近缘种相比,生活在高原的动物通常需要更多的能量来适应高原缺氧、低温、强紫外线的极端气候。其中,瘤胃肠道微生物群中的碳水化合物活性酶(CAZymes)不仅参与多糖和单糖的转化,对SCFAs/VFAs的产生也有重要贡献。已有研究指出,高原有蹄类动物如藏羚、藏野驴和野牦牛相对于其它平原近缘种,其肠道微生物具有较高的SCFAs合成效率,可能归因于它们独特的肠道微生物群落组成,但具体的遗传特征尚不明确。
猜 想
与平原近缘种相比,高原有蹄类动物肠道微生物群中观察到的高SCFA/VFA生产效率表型,可能与它们一些核心的SCFA/VFA产生肠道微生物群物种中较高CAZyme丰度的趋同进化有关。具体来说与三个潜在因素有关:第一,高原宿主中一些具有较高CAZyme活性的核心肠道微生物群丰度较高;第二,高原宿主中一些核心肠道微生物群存在具有较高CAZyme编码基因的适应性基因组结构变异(SVs);第三,高原宿主通过其他具有较高CAZyme编码基因的进化和物种形成模式建立了独特的肠道微生物群。
实验设计
研究背景
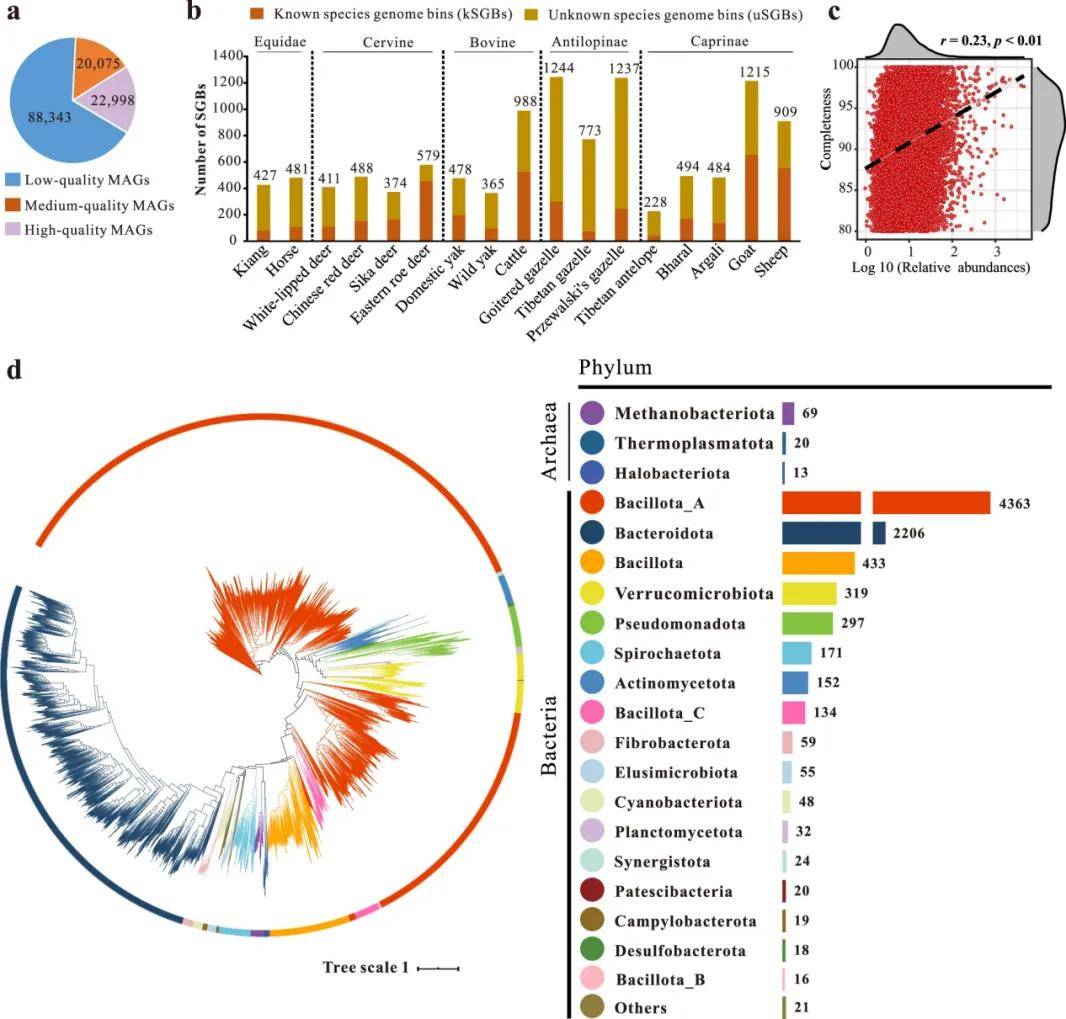
图2 | 17种有蹄类动物肠道微生物群中的基因组组装结果
17 种常见有蹄类动物的分布及生存状况
针对去除宿主后的高质量序列进行组装和分类,生成131,416个MAGs。按照完整性和污染度划分为88,343个低质量MAGs(完整性<50%或污染度≥5%)、20,075个中等质量MAGs(完整性≥50%且污染度<5%)和22,998个高质量MAGs(完整性≥80%且污染度<5%)(图2a)。随后使用spearman相关性系数评估MAGs丰度与完整性之间的相关性。结果显示,MAGs丰度与完整性之间呈正相关关系(图2c)。
使用dRep分别在菌株水平和种水平对每种有蹄类动物肠道微生物群中的高质量MAGs进行去重,对获得的SGBs根据GTDB-Tk的分类进一步划分已知SGBs(kSGBs)和未知SGBs(uSGBs)(图2b),并将合并后的全部SGBs去冗余(ANI≤95%),最终得到一组8489个非冗余物种级基因组箱SGBs。这些非冗余SGBs涵盖2个界、29个门、42个纲、102个目、230个科和1061个属。其中,界水平主要为细菌(8387 SGBs),门水平主要为厚壁菌门(Bacillota,4363 SGBs)和拟杆菌门(Bacteroidota,2206 SGBs)(图2d)。
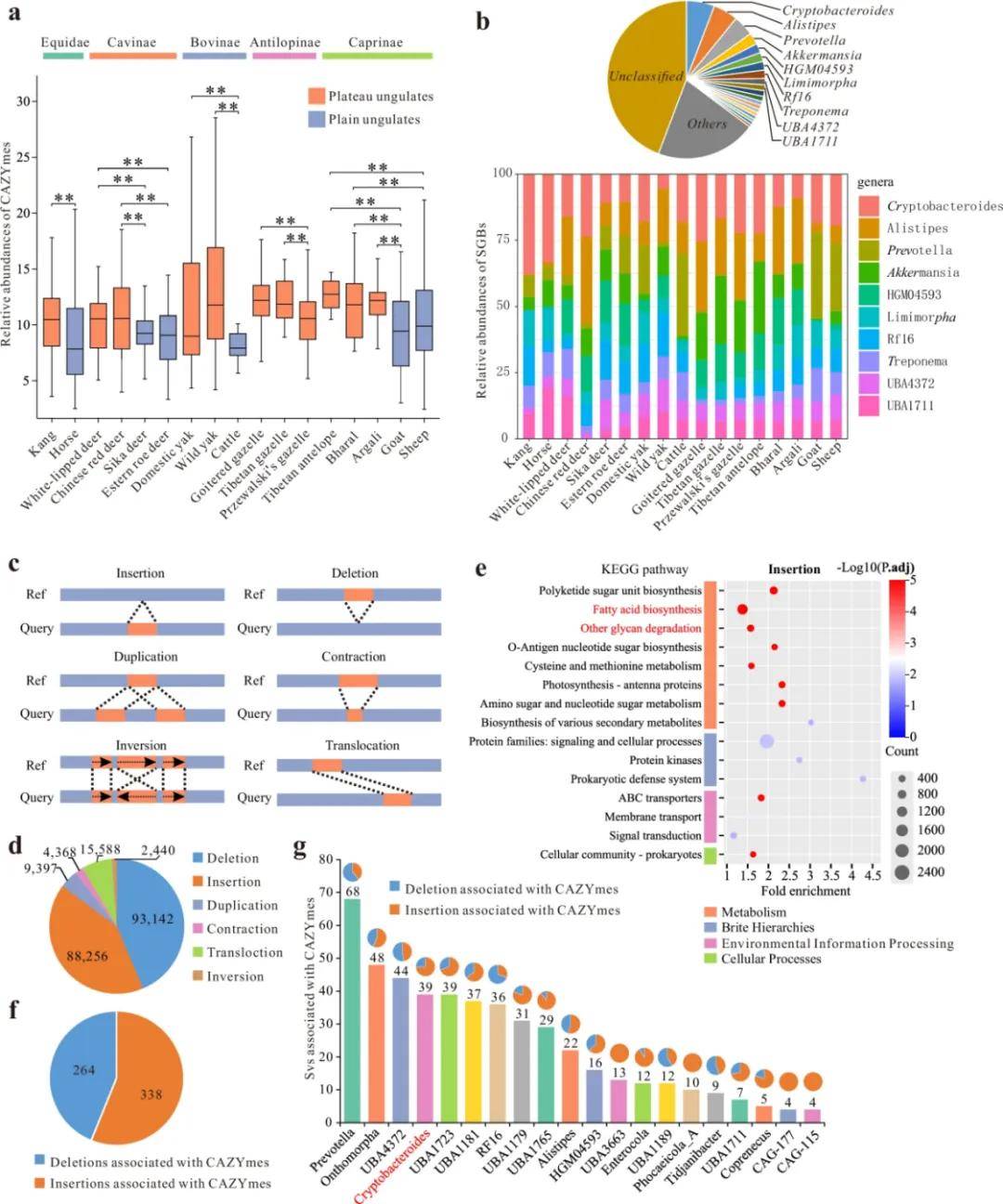
图3 | 高原和平原有蹄类动物MAGs中CAZYmes和结构变异(SVs)的验证、特征描述及功能相关性
高原有蹄类动物肠道微生物组结构变异SVs与CAZymes基因丰度升高的关系
使用dbCAN2注释contigs中的CAZymes并统计非冗余相对基因丰度。从结果看,高原有蹄类动物肠道微生物组中CAZymes的相对丰度均高于其它低地同类(图3a)。
然后注释所有11,175个SGBs中的CAZymes,发现在高原宿主中,SGB相对丰度和CAZyme基因密度均最高的属(如隐杆菌属和普氏菌属),其比例并非总是高于平原近种(图3b)。因此,高原有蹄类动物肠道中产SCFA/VFA且具较高CAZyme基因密度的丰度较高的微生物群可能并不是高效产生 SCFA/VFA 表型的主导因素。
为了进一步研究高原宿主肠道微生物组中CAZymes基因丰度较高的可能因素,通过比较上述过程中生成的每个物种簇中的MAGs来检测结构变异(SVs),包括插入、缺失、重复、易位等(图3c)。鉴定结果显示84.98%的SVs为插入(88,256个;41.35%)和缺失(93,142个;43.63%)(图3d),这可能是导致肠道微生物组功能基因得失的因素之一。
基于这部分数据,利用KEGG数据库对与插入和缺失相关的功能基因进行通路富集分析,并分别分析受影响的top15通路(图3e),发现受影响最大的KEGG通路主要与能量代谢相关(8个通路),如“脂肪酸生物合成”、“聚酮糖单元生物合成”等(图3e);其他通路主要涉及分子运输、细胞防御和信号转导(图3e)。但缺失SVs受影响最大的KEGG通路并未呈现出与插入SVs中相似的富集模式。
已有研究表明,微生物组基因组中的SVs通常与CAZYme等功能基因的获得和丢失有关,所以接下来对CAZymes相关的插入和删除进行研究。数据显示对于高原和低地宿主中普遍存在的肠道菌群菌株(特别是隐杆菌属),由于其基因组中存在插入结构变异,高原个体的基因组比低地个体获得更多的CAZymes。
最后,我们使用gutSMASH将所有与插入和缺失相关的功能基因(包括CAZymes)重新映射到一个包含41种不同已知通路的数据库中,以分析涉及的主要代谢基因簇(MGCs),并选取瘤胃肠道微生物组中参与SCFAs/VFAs生产的14种关键酶进行分析,结果显示与平原有蹄类动物相比,高原有蹄类动物中大多数用于SCFAs/VFAs生产的关键酶表现出更高丰度。
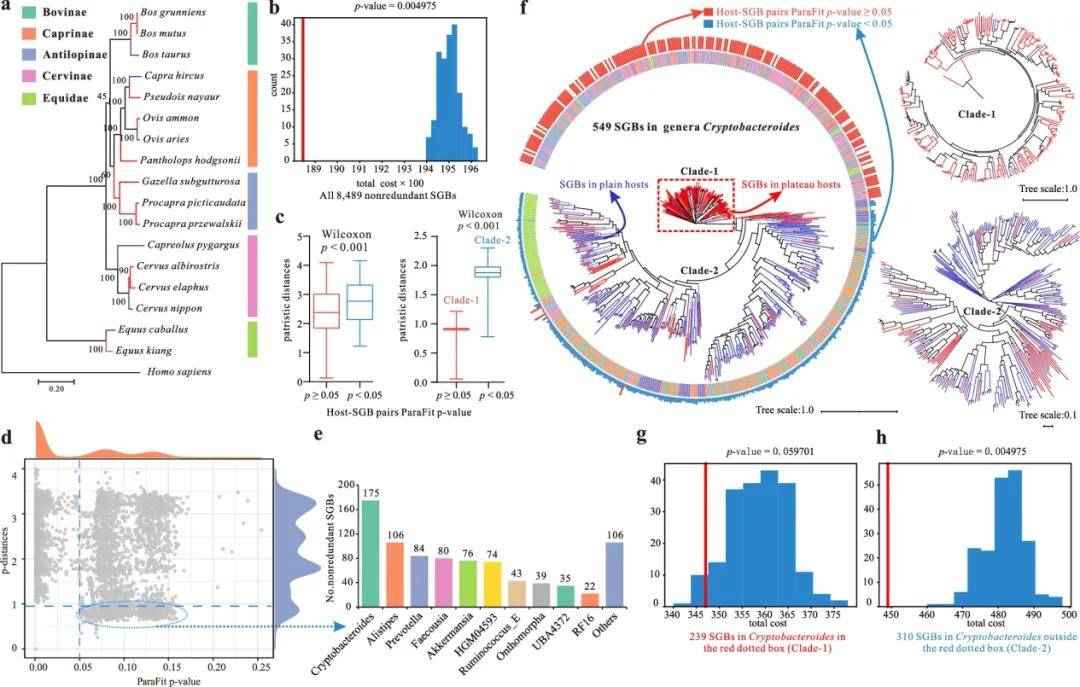
图4 | 肠道微生物组与其有蹄类宿主的协同进化关系
高原有蹄类动物肠道微生物群的广泛独立快速分化
除了在高原和平原有蹄类动物中普遍存在的肠道菌群在能量代谢方面表现出基因组差异外,我们还推断高原有蹄类动物中可能存在一些独特的肠道菌群,它们也可能具有适应性的基因组特征。
故先构建17种有蹄类动物的最大似然树(图4a),并计算肠道微生物群及其宿主的寄生距离矩阵,此外,eMPRess软件在宿主 - SGB关系中于0.01水平上否定了随机形成的零假设(p = 0.004975)(图4b)。因此从整体看,这些非冗余SGBs与其宿主是协同进化的。
接着ParaFit H - S链接模型测试结果表明,5095个非冗余SGBs与其宿主表现出显著的协同进化关系,而其余的3394个非冗余的sgb与它们的宿主没有共同进化,更重要的是,在未协同进化的非冗余SGBs中,大部分都属于隐杆菌属(图4d、e)。因此,大量来自高原宿主的未协同进化的物种水平基因组分箱(SGBs)被识别出来,并且一些肠道微生物群落正在经历独立的快速进化和物种形成过程。其中隐杆菌属的肠道微生物组不仅表现出导致获取更多CAZymes的结构变异,而且还呈现出独特的进化和物种形成模式。因此,对该属的所有SGBs的系统发育树进行了重建和可视化(图4f和补充图7),从隐杆菌属的树中观察到,红点框内的SGBs(归为分支1)主要来自高原宿主,并且与红点框外的SGBs(归为分支2)相比,其分支长度更短(图4c、f)。
此外,分别使用ParaFit、PACo和eMPRess确认了SGBs与其宿主之间的协同进化关系。具体如下:首先使用ParaFit H - S链接模型确定隐杆菌属中每个H - S链接的p值(图4g),分支1中每个H - S链接的p值通常≥0.05,而分支2中通常 < 0.05(图4f)。随后,基于该系统发育树,对分支1和分支2中的SGBs到根物种水平基因组分箱分支(EQK.GHM.031_bin.100)的平均相对p-distance进行分析,结果显示分支1中SGBs的相对平均p -distance明显比分支2中更近,与所有SGBs的分析结果一致(图4c)。接着分别重建分支1和分支2中SGBs的系统发育关系(图4f),在分支2中检测到了协同进化关系(图4h),但在分支1中未检测到(图4g)。也就是说,在高原有蹄类动物的肠道微生物组中,尤其是在隐杆菌属(Cryptobacteroides)的SGBs中,检测到了广泛的独立快速物种形成现象。
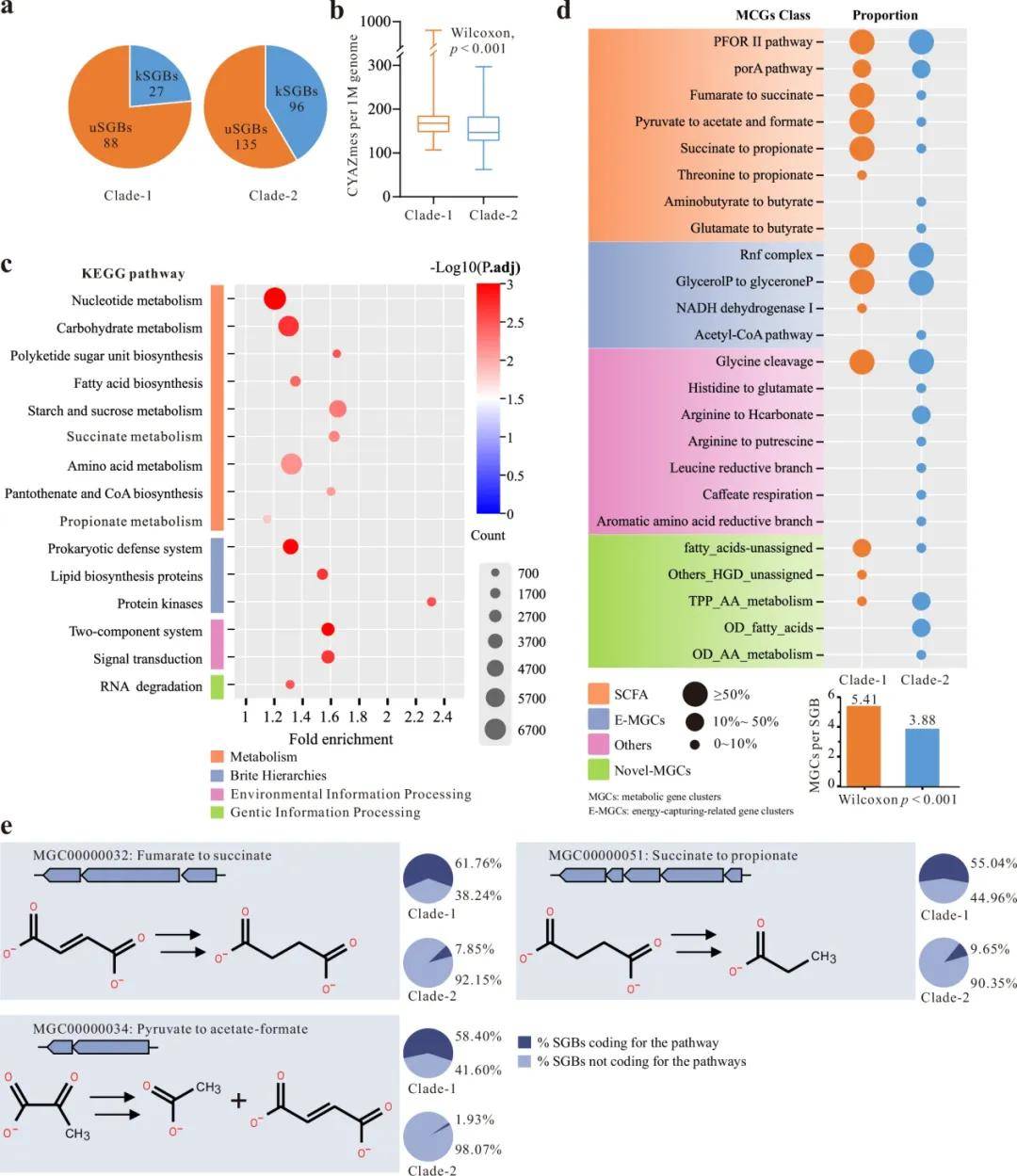
图5 | 分支1和分支2的SGBs中的基因组特征及初级代谢基因簇(MGCs)
隐杆菌属分支1中的新型肠道微生物组物种在短链脂肪酸产生方面显示出更高的潜在能力
为了深入研究隐杆菌属分支1内SGBs的基因组特征,重点研究了去重后生成346个非冗余SGBs。根据GTDB-TK的注释,分支1中76.52%的SGBs被归类为未知SGBs,这一比例高于分支2(58.44%;图5a)。随后CAZymes注释结果显示分支1中CAZymes的基因密度显著高于分支2(图5b)。随后基于KEGG数据库对SGBs内的基因进行注释,并筛选分支1中比分支2丰度更高和分支2中比分支1丰度更高的前5%基因进行KEGG通路功能富集分析(图5c)。结果显示,分支1中比分支2丰度更高且具相同ko编号的基因主要富集在能量代谢通路中,如“碳水化合物代谢”、“脂肪酸生物合成”等(图5c)。然而,在分支2中比分支1丰度更高的基因,并未观察到类似模式。
使用gutSMASH检测分支1和分支2中的主要代谢基因簇(MGCs)。结果显示,分支1中MGCs的比例(每个SGB 5.41个MGCs)均高于分支2(每个SGB 3.88个MGCs;图5d)。具体来说,分支1和分支2中的MGCs都聚集在与丙酮酸异化以及乙酸和丁酸生成相关的SCFA通路中,例如“PROR II pathway”和”porA pathway”(图5d)。分支1中编码诸如”fumarate to succinate”, “pyruvate to acetate and formate”, 和 “succinate to propionate” 等通路的SGBs比例高于分支2,这意味着分支1肠道微生物组中短链脂肪酸(此处为乙酸、甲酸和丙酸)的生产效率更高(图5d、e)。
研究结论
本研究创建了一份关于中国常见有蹄类动物肠道微生物群的全面目录。基于大规模、高深度的宏基因组测序,实验观察到高原有蹄类动物肠道微生物群中碳水化合物活性酶(CAZymes)的高丰度广泛趋同进化现象,与之相关的两个主要因素:(1)基因组结构变异导致高原有蹄类动物核心肠道微生物群中CAZyme基因增加(2)独特的肠道微生物物种,特别是隐杆菌属,由于经历快速的独立进化和物种形成,导致更高的CAZyme基因密度。
总体来说,本文的发现为肠道微生物组的基因组变异、进化和物种分化提供了新见解,加深了我们对宿主及其肠道微生物组之间共同适应的理解。
原文引用
Xu B, Song P, Jiang F, et al. Large-scale metagenomic assembly provide new insights into the genetic evolution of gut microbiomes in plateau ungulates[J]. npj Biofilms and Microbiomes, 2024, 10(1): 120.
原文链接
https://doi.org/10.1038/s41522-024-00597-3


















