

2025-01-06

前言
染色体外环状DNA(eccDNA)是一类游离于染色体外的环状DNA,广泛存在于包括人类在内的多种真核生物中,并参与基因表达调控、细胞增殖、DNA损伤修复及肿瘤发生等多个生物学过程。由于eccDNA不易被核酸酶降解,且结构较为稳定,因此具有重要的生物学功能。目前,研究eccDNA的主要方法包括DNA荧光原位杂交(FISH)、批量全基因组测序(WGS)和Circle-Seq。前两种方法主要用于检测来源于相对较大基因组区域、丰度较高的eccDNA,而Circle-Seq则能够识别广泛的eccDNA,但需要较大数量的细胞进行测序分析。因此,亟需开发一种通用且可扩展的方法,用以检测从组织活检中提取的单个细胞或细胞核中的eccDNA,从而揭示eccDNA的生物学复杂性,并阐明其在患者来源肿瘤样本中的异质性。
2024年2月,瑞典卡罗林斯卡医学院的科研人员在Nature Communications杂志在线发表题为“scCircle-seq unveils the diversity and complexity of extrachromosomal circular DNAs in single cells”的研究论文。研究团队开发并验证了Circle-seq的单细胞应用scCircle-seq,该方法适用于非固定和固定的细胞或细胞核,包括从肿瘤活检组织中提取的细胞核。研究显示,大多数eccDNA在同一类型的细胞之间表现出高度的可变性,且倾向于在基因组扩增区域形成。此外,将scCircle-seq应用于三种不同癌症类型的肿瘤样本中,研究人员发现了肿瘤特异性的eccDNA图谱,并揭示了具有不同eccDNA基因组模式的亚克隆群体。综上所述,scCircle-seq作为一种易于扩展、直接且通用的方法,具有揭示癌症中eccDNA生物学复杂性和异质性的潜力。
01、技术原理
为了开发scCircle-seq,研究团队在Circle-Seq流程的基础上引入了一个额外的DNA缺口修复步骤,以提高eccDNA的检测效率,并设计了一个通用的工作流程,适用于通过多孔板或单管分选的活细胞、固定细胞以及细胞核。为了解析scCircle-seq数据,研究团队改编了先前用于分析大规模Circle-Seq数据的生物信息学流程:该流程首先识别具有高测序覆盖度的基因组区域(即产生eccDNA的基因组区域,CPRs),然后通过搜索映射在CPR内的过度表示的非一致性和分裂reads对,确定每个CPR内的嵌合连接。此外,研究团队还验证了scCircle-seq的特异性,结果与预期一致。
为了进一步验证该方法,研究团队将scCircle-seq应用于5个不同的细胞系,包括4个癌症衍生细胞系(HeLa、K562、Colo320DM和PC3)和1个永生化正常细胞系(293T)。结果表明,鉴定出的CPRs数量及其对应的基因组覆盖率在单个细胞和细胞系之间存在显著差异,这与已发表的Circle-Seq数据一致,表明大多数eccDNA在细胞间表现出显著的差异,并且在细胞分裂过程中是以随机方式遗传的。
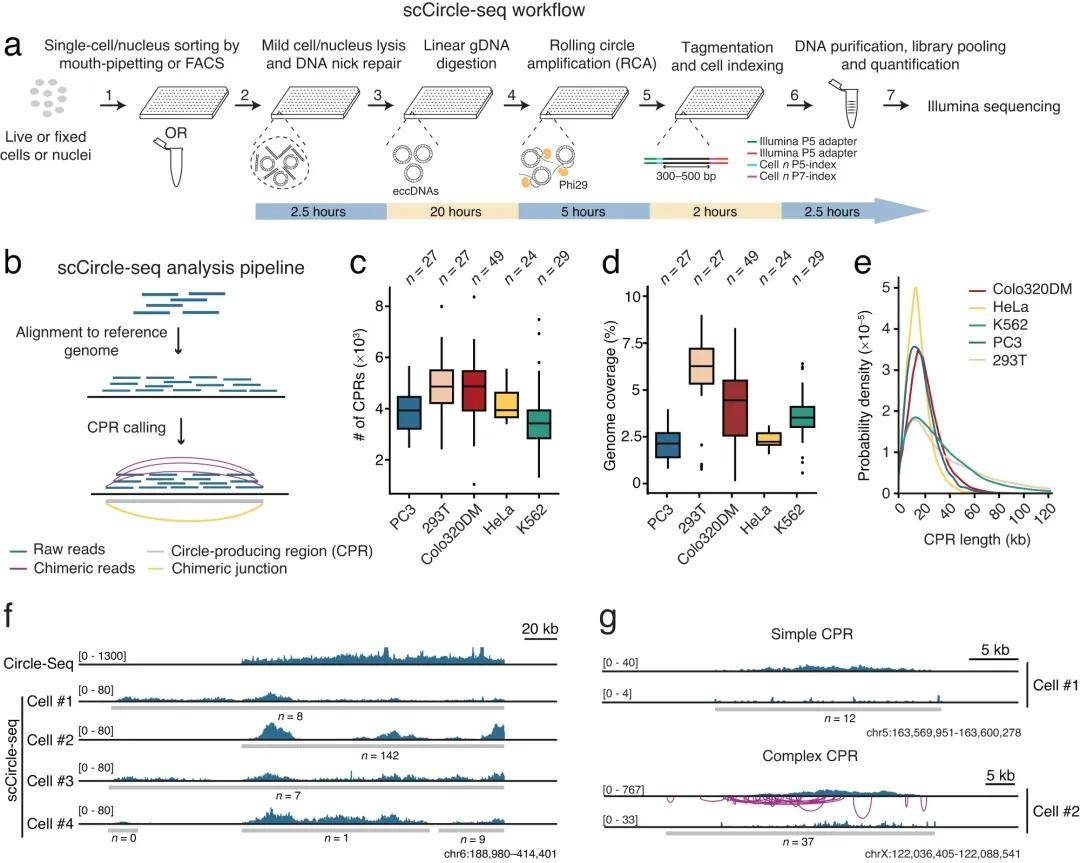 图1 scCircle-seq工作流程及数据验证
图1 scCircle-seq工作流程及数据验证
02、研究内容
1、单细胞中eccDNA的基因组图谱
研究团队分析了scCircle-seq检测到的eccDNA的基因组分布,以确定是否存在特定的模式。结果发现,eccDNA在单细胞水平上表现出极大的变异性,但在群体水平上可能呈现出更明确的分布模式。根据同一细胞系中所有单细胞中对应CPR的频率及其均匀性评分,研究团队将scCircle-seq鉴定出的eccDNA分为四组进行验证(见图2)。结果显示,绝大多数检测到的eccDNA(88-99%)被归类为低频低均匀性(LFLU),少数则归类为高频高均匀性(HFHU),主要存在于Colo320DM和293T细胞中。上述结果表明,eccDNA本质上具有高度的异质性,但scCircle-seq能够有效区分不同类型的eccDNA。
进一步的分析将鉴定出的CPRs与各种基因组注释数据进行交集,探索了scCircle-seq检测到的eccDNA在基因组中的分布。结果发现,CPRs在组蛋白H3K9me3(构成性异染色质)和H3K27me3(选择性异染色质)标记的染色质区域中富集,表明eccDNA的形成主要集中在异染色质区域。
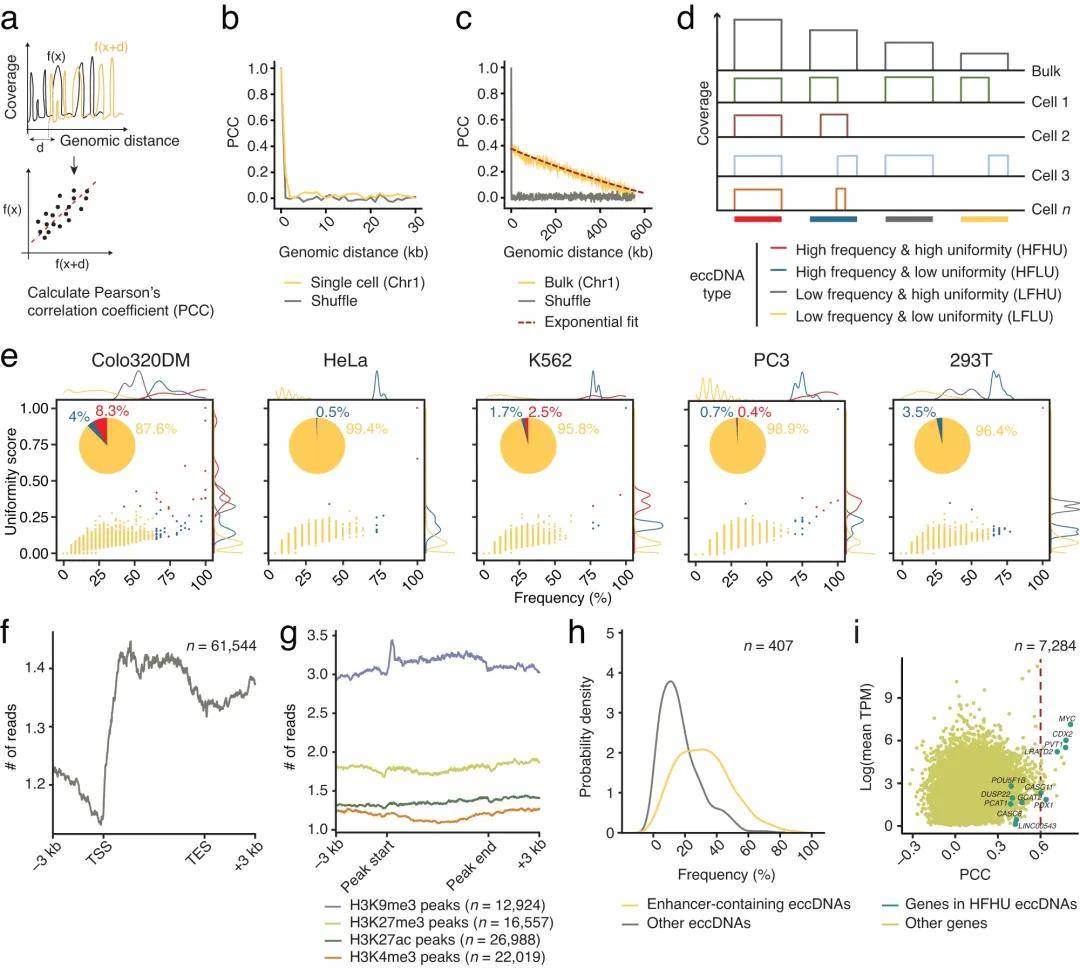 图2 scCircle-seq揭示了单细胞中染色体外环状DNA的多样性和复杂性
图2 scCircle-seq揭示了单细胞中染色体外环状DNA的多样性和复杂性
2、基于eccDNA的细胞类型分类
进一步,研究团队探讨了细胞中的eccDNA序列是否呈随机分布,或者是否能够利用scCircle-seq识别不同细胞类型的eccDNA特征(见图3)。首先,研究团队将scCircle-seq检测到的CPRs基因组分布表示为一个“cells × bins”矩阵,其中“cells”表示通过scCircle-seq分析的单个细胞数量,“bins”则是定义好的长度的连续基因组窗口。接着,团队使用cisTopic计算框架对细胞进行聚类,并根据可用的基因组轨迹对每个聚类进行注释。
结果显示,该方法能够成功地将同一细胞类型的细胞聚类在一起。进一步分析表明,尽管相同类型细胞之间的eccDNA数量和来源区域存在较大差异,不同细胞类型仍然表现出一定的特定eccDNA特征,这些特征部分反映了各自的表观基因组特征。
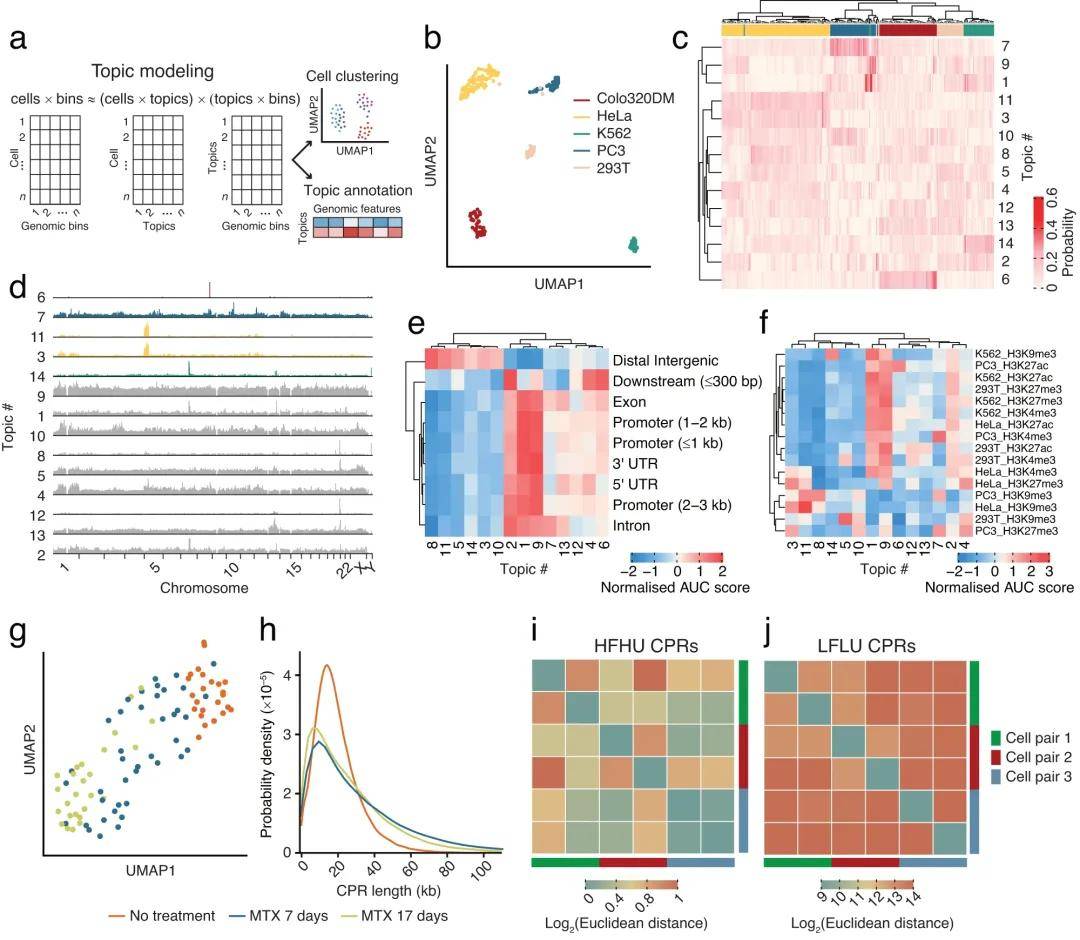 图3 eccDNA的细胞特异性和动力学
图3 eccDNA的细胞特异性和动力学
3、scCircle-seq应用于患者来源肿瘤样本
研究团队将scCircle-seq应用于从三种回顾性收集的冷冻肿瘤样本中提取的细胞核(分别为1例前列腺腺癌PRAD、1例三阴性乳腺癌TNBC和1例管腔B型乳腺癌LumB),验证了scCircle-seq在患者来源肿瘤样本中的适用性。最终,研究团队获得了55个PRAD细胞核、87个TNBC细胞核和33个LumB细胞核的高质量测序数据,所有细胞中的环形DNA与线性DNA的比值接近100%,这一结果与从永生化细胞系中获得的结果一致,表明即使在肿瘤活检中提取的细胞核,scCircle-seq仍能稳定有效地工作。
结果显示,PRAD细胞中的CPRs数量最多,且覆盖的基因组区域比例最高,其次是TNBC和LumB细胞,表明这些肿瘤细胞具有不同的eccDNA图谱。此外,研究团队对单细胞CPRs的基因组图谱进行了UMAP降维分析和差异主题分析,发现PRAD与TNBC、LumB细胞形成了两个明显不同的簇,表明这两类肿瘤携带不同的eccDNA基因组特征,这与它们各自的起源组织一致。
进一步,研究团队将LumB和TNBC细胞中鉴定的CPRs与癌症基因组图谱(TCGA)数据库中乳腺癌样本的体细胞拷贝数变化(SCNAs)数据进行了交叉分析。结果显示,相较于TNBC簇2细胞的CPRs,TNBC簇1和LumB细胞的CPRs在扩增区域中显著富集。研究团队还分析了鉴定的eccDNA数量与相应基因组区域拷贝数之间的关系,发现CPRs的数量在二倍体和中度扩增区域之间无显著差异,而在扩增水平较高的区域则明显增多。上述结果表明,肿瘤细胞的eccDNA分布反映了其SCNA特征,高度扩增的基因组区域更容易产生更多的eccDNA。
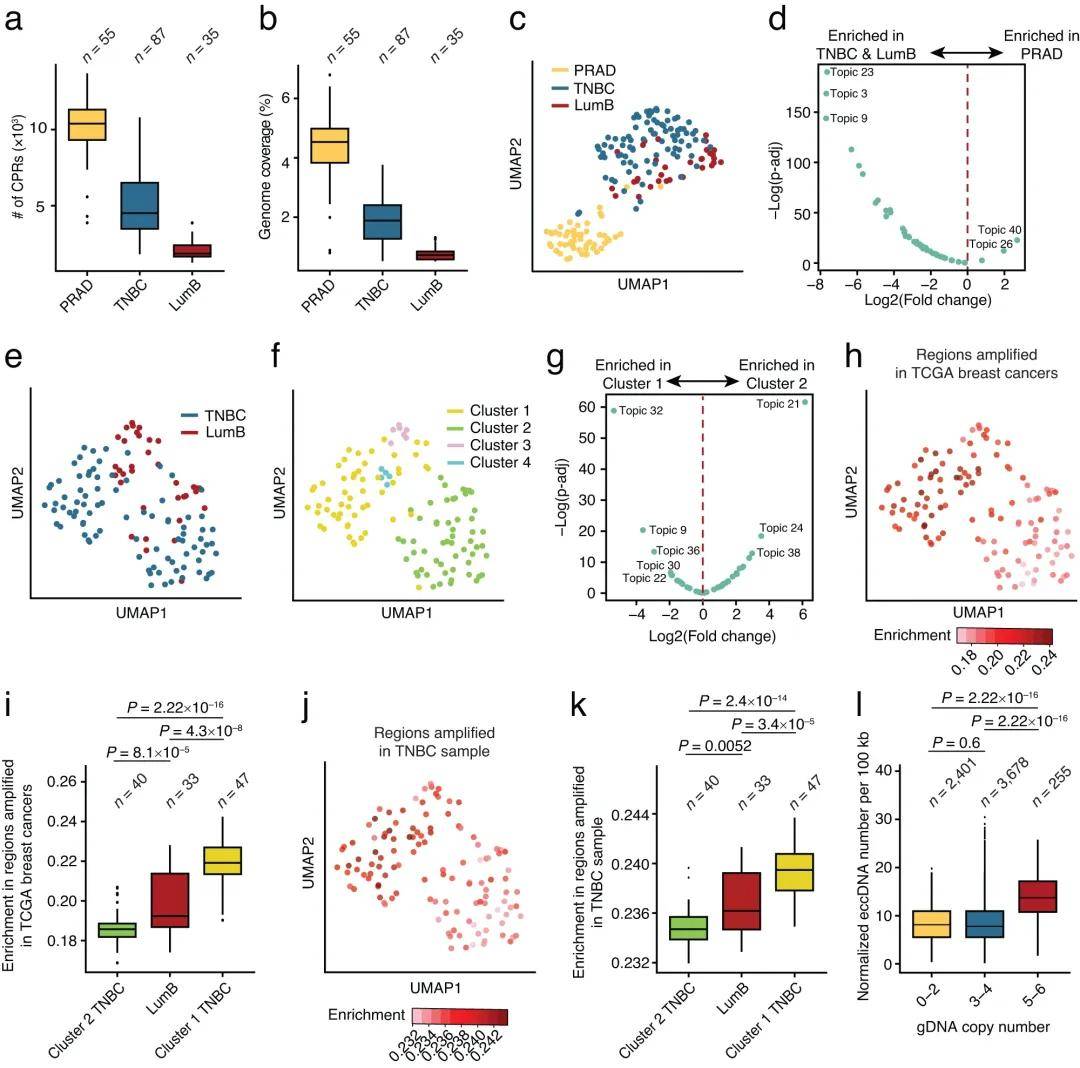 图4 scCircle-seq揭示了单细胞中染色体外环状DNA的多样性和复杂性
图4 scCircle-seq揭示了单细胞中染色体外环状DNA的多样性和复杂性
03、总结
综上所述,研究团队开发了scCircle-seq方法,能够在单细胞水平上深入分析eccDNA的多样性和复杂性。scCircle-seq结合了滚环扩增(RCA)和DNA缺口修复步骤,显著提高了检测灵敏度,不仅能够识别富集的、含有癌基因的eccDNA,还能检测到罕见的eccDNA。特别地,scCircle-seq简化了工作流程,降低了低丰度eccDNA丢失的风险。总体而言,scCircle-seq是一种可扩展的工具,适用于分析不同细胞和组织类型中的eccDNA复杂性,并有望进一步推动eccDNA在癌症诊断中的应用潜力。


















