

2025-01-08

denovo测序常见问题
Q1: denovo基因组组装一定要做Survey?
对于没有参考基因组的物种,在启动denovo项目之前,对基因组特征评估是十分必要的,基因组大小及复杂状况直接影响到基因组组装策略、测序数据量、完成周期等。
Q2:怎样评估研究物种的基因组大小?
(1) 网站查询:查询植物基因组大小的网站:http://data.kew.org/cvalues;查询动物基因组大小的网站:http://www.genomesize.com;
(2) 流式细胞仪方法:流式细胞仪是目前比较常用的估计基因组大小的实验方法;
(3) Survey评估:即将测序得到的reads打断成K-mer,通过K-mer分析,从数学的角度评估基因组的大小,杂合以及重复等信息。并进行初步组装,从初步组装的Contig的GC分布图上,判断该物种是否有污染等信息,从而为后续组装策略的制定提供可靠的依据。
Q3:如何保证组装结果的可靠性?
对于组装的结果,除了保证Contig N50和Scaffold N50两项指标外,还需要对组装质量进行评估。如利用EST数据和 RNA 数据进行完整性评估,即评估组装出来的基因的完整性;利用BAC数据检验是否有装断或装错的情况,以及利用保守基因评估(CEGAM评估/BUSCO评估)基因组组装的完整性。
Q4:Survey 和基因组denovo所用DNA是否需要一样的?
原则上进行Survey和denovo使用的DNA是来自一个个体的。如果DNA量不足以满足整个denovo项目,则建议小片段文库的DNA必须来自同一个体,大片段文库使用同一群体的另一个个体。
Q5:denovo分析的内容有哪些?
(1) 基因组概况:重复序列、转座子、编码基因、非编码 RNA;
(2) 比较基因组学(包括基因家族分析:venn图、基因家族扩张/收缩、扩张基因家族功能富集分析;共线性分析;单拷贝基因蛋白序列一致性)
(3) 系统演化分析(包括系统发育地位、有效群体大小估计、物种分化时间估算、全基因组复制分析 (WGD)、新物种鉴定)
(4) 物种特异性分析(例如榴莲挥发性硫化物分析、大熊猫小熊猫协同进化分析等)。
BSA常见问题
Q1:做BSA需要哪些准备?
(1) 性状差异明显的亲本;
(2) 分离群体(F2、RIL、BC 等)中能够准确的选出极端个体(表型鉴定一定要准确)构建混池;
(3) 有合适的参考基因组信息。
Q2:做 BSA 适合的群体类型有哪些?
遗传群体的构建类型有很多种,其中F2、BC、RIL为常见的可做BSA的群体,DH也可⽤于BSA性状定位,但由于构建困难,不常见。RIL、DH等永久性分离群体优于F2、BC等暂时性分离群体。此外,F1代仅用于林木或鱼类等亲本杂合度较高的物种中基因的定位。用F1群体进行BSA分析风险较高,可能出现较多的假阳性,甚至会出现无法定位出候选区域的情况。
Q3:BSA样品准备时,DNA量需要多少,有什么质量要求?
每个样本DNA要求:每次建库需要准备样品2μg,请提供2次制备的量。样品浓度>20ng/μl;OD260/280 介于1.8-2.0,无肉眼可见污染;基因组完整、无降解,电泳中DNA主带应大于23kb。样品选择:对于植物样品建议选取黑暗培养的黄化苗或嫩苗;动物样品应选择肌肉、血等脂肪含量较少的组织进行取样。
Q4:只测 2 个子代池是否可行?
可以,但容易产生假阳性位点。如果研究性状为EMS诱变的质量性状,同时所研究物种已有组装质量较好的参考基因组,研究品系亲本为普通野生型,与参考基因组品系为相同生态型的情况下,是可以只测2个子代池的。但是一般情况下,由于参考基因组和所研究品种之间原本就可能存在较大的差异,会导致出现大量假阳性SNP,还是推荐对2个亲本和 2 个子代池都进行测序,尽可能排除背景噪声,减少假阳性位点。
Q5:如何确定子代池样本数量以及测序深度?
在《User guide for mapping-by-sequencing in Arabidopsis》中,作者对混池个体数以及测序深度对候选基因数量的影响进行了评估。结果显示,当子代个数超过30个,测序深度大于30X之后,定位出候选基因的数量趋于稳定。同时基于多篇文献的报导,推荐个体数≥ 30 个/池,测序深度:每个亲本≥ 20X,每个子代池≥ 30X。
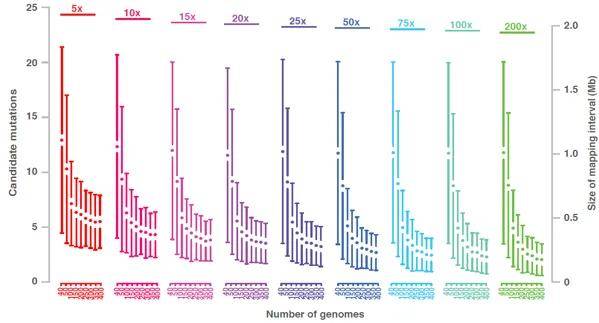
Q6:候选基因如何确定?
在整个基因组范围内挑选候选SNP和InDel,挑选⼦代池中SNP-index接近0的位点或者接近1的位点作为SNP、InDel候选位点。根据候选位点的注释信息,优先挑选引起⾮同义突变或者可变剪接位点或者移码突变的位点,这些位点所在的基因作为候选基因。
Q7:定位到的候选区间很大(大于几兆),或出现多个候选区间,怎么办?
没测亲本、目标性状考察不准确或者目标性状是由多基因或微效QTL控制的数量性状都会导致该结果。常见筛选⽅法如下:
1)利用marker assistant SSR/InDel/SNP等分子标记辅助筛选,利用双亲或两个混池个体间基因型的多态性将候选区间范围进一步缩小;
2)根据基因功能注释结果筛选,找到的几十个基因不一定都和研究的性状相关,去除无关基因;
3)结合已发表的遗传图谱QTL定位的结果,将候选区间缩小;
4)构建更大的群体进行定位,增加取样数,增加测序深度;
5)通过多组重复实验,取交集。
群体进化常见问题
Q1:群体进化研究的群体选择标准?
对所研究物种的各个亚群,要求尽量亚群间划分明显,同一亚群内的个体要有一定代表性。如研究驯化机制,则需要选取野生型、驯化型样本;如研究适应性进化,则需要选取不同地理环境、不同海拔高度的样本;如研究种群历史,需要在物种的可能起源地以及各个分布区域进行选材。
Q2:群体大小选择有什么要求?
通常至少需要三个亚群/亚种,每个亚群选取10个样本左右(推荐动物≥10个,植物≥ 15个,珍稀物种可适当减少个体),总体建议不少于 30 个样本。
Q3:每个群体取 10 个样本与每个群体取 15 个样本的区别有多大?
一般认为群体的个数越多越能代表这个群体的全部信息。15 个样本的群体所代表的群体信息肯定是大于10个样品。综合考虑项目成本和分析效果,建议动物取10个以上,植物取15个以上,这样的性价比应该是最高的。
Q4:针对群体遗传进化,测序深度一般有什么要求?
综合考虑项目成本及结果准确性,建议群体进化研究的测序深度不低于10X。随着测序成本降低及分析方法成熟,高深度测序应用越来越普遍:对于某些亚群的关键个体可以进行 50X高深度测序,建立该群体高质量变异数据库,进行CNV等大结构变异检测及差异比较;或者对群体的每个个体加深测序深度进行基于CNV的选择消除分析等,来挖掘大结构变异导致的进化及性状形成机制。
Q5:群体进化分析,需要知道哪些样品信息?
需要知道目标物种有无参考基因组,样本分为哪些群体(种/亚种/变种/变型/居群/品种/品系);主要研究目的(适应性研究/种群历史研究/种群迁移研究/种间杂交研究等)。此外还有样品取样地信息(经纬度、海拔、地区等)。在进行选择性消除时,可能需要提供样品的表型性状信息。
Q6:如何理解选择性清除分析?
选择消除现象(Selective sweep)是由于某一点受到强选择后,其周围位点的多态性因受该位点牵连而发生多态性降低的现象。当某个位点发生突变,突变后的位点因对物种在特定的情况下有利或者受到了人为的选择,那么该突变位点在群体中的频率必然提高,但是其附近和它处在同一个单体型或者block的其他多态性位点同样跟着受到了选择,频率提高,也就是该单体型内其他多态位点的某一多肽型大大提高,从而降低了周围区域的多态性。
Q7:群体进化怎样与其他项目联合分析?
群体进化可以与全基因组关联分析结合,研究进化中受选择位点与表型性状的关联;群体进化与地质环境分析结合,研究种群演化历史与古气候变化的关系;群体进化与比较基因组学分析结合,从宏观和微观的尺度研究物种形成及其种群分化。
遗传图谱常见问题
Q1:影响遗传图谱上图标记数的因素有哪些?
遗传图谱项目最终上图标记数受各种因素影响,如基因组大小,测序策略,分离群体类型,亲本纯合度,SNP 密度,SNP 标记过滤参数等。根据相应的测序方案和过滤标准,可以归结为以下几点:
(1) 亲本间多态性 SNP 数量(即要求亲本间要有一定的多态性);
(2) 符合分离群体类型的标记比例(与亲本纯合度有关):BC1、F2、RILs 分离群体多在 70% 以上;高杂合的亲本(纯合度低于 50% 的亲本)一般用 F1 分离群体构图;
(3) SNP 过滤标准:一般为SNP的缺失率不大于25%,偏分离检测 p<0.05,标记质量一般大于5X。过滤标准可以根据项目具体情况调节。
Q2:构建遗传图谱,如何选择作图亲本及所需群体大小?
首先,亲本间基因组多态性要高,一般异交作物的多态性高于自交作物;其次,亲本尽可能为纯度高的材料,若亲本杂合度高(如林木类),要采用F1群体构图方案;最后,亲本杂交后代需具有可育性,并且群体大小决定着遗传图谱的检测效率,群体越大,检测效果越好。一般情况,样本数≥200个时,能构建较为精确的遗传图谱。
Q3:构建遗传图谱,对亲本及子代测序深度有何要求?
参考测序深度是衡量分子标记可靠性的一个标准。一般情况下,亲本推荐各测10-20×,子代测序深度与测序方式有关,一般重测序要求5X以上。
Q4:亲本和参考基因组是一个品种,是否需要重新对亲本进行测序?
需要。即使是同一个品种,不同来源的种质可能存在很多未知的差异。如水稻品种9311,不同来源的 9311 表现出明显不一致的表型,比如有的有芒,有的无芒。所以这种情况下,建议老师重测,但是测序深度可以稍微降低些。
Q5:遗传图谱中的QTL定位与BSA定位,二者区别是什么?
遗传图谱需要对每个个体测序,可以检测到所有的QTL位点;BSA是挑选极端性状的样本进行混池测序,仅能检测到主效的QTL位点。
Q6:用于QTL定位的表型数据是否必须满足正态分布吗?
大部分QTL定位方法只是要求表型数据中的随机误差项服从正态分布,并没有要求表型数据满足正态分布。数量性状只有在多基因假说下才真正符合正态分布,一般情况下,表型数据呈非正态分布并不影响QTL定位。
GWAS常见问题
Q1:对于多年多点重复实验材料,需要提取所有材料的DNA吗?
对于作物材料存在自交种和杂交种:(1)自交种一般接近纯合,已经非常稳定,一个样本就可以代表一个品种的遗传信息,所以自交种的样本只需选择一个地点的一株材料进行 DNA提取即可;(2)杂交种也是如此,品种都具有特异性 (Distinctness)、一致性 (Uniformity) 和稳定性(Stability),所选的材料如果是品种,则具有一致性和稳定性,即一株材料可以代表该品种的遗传信息,所以杂交种也只需选择提取一个地点的一株材料进行DNA提取即可。
Q2:GWAS所需的最低样本数和测序深度是多少?
全基因组关联分析主要针对的是自然群体,群体越大,检测效率越高。目前 GWAS 产品建议样本数在200个以上,测序深度至少10X以上。
Q3:GWAS分析对表型选择有什么要求?
选择遗传力较高的表型,遗传度低的表型会降低遗传学关联研究的准确度。性状优于疾病(表型):疾病(表型)的状态模糊不清,很难测量,有时则会出现多种疾病(表型)混杂在一起而难以判断。数量性状要选择测量简单准确并且遗传力相对较高的,以增加分析结果的可信度。
Q4:如何降低分析结果的假阳性?
在全基因组关联分析中,前期对样本的采集情况(表型分布均匀,环境一致)会对后续分析的假阳性结果存在最大的影响,在分析过程中会采用如下方法降低分析结果的假阳性:
(1) 结合群体分层信息,利用混合线性模型,对结果进行校正;
(2) 必要时还会采取多种线性模型进行分析降低假阳性;
(3) 采用Bonferroni校正法来校正GWAS分析中多重假设检验后的 P 值以降低假阳性的概率。
Q5:研究不同性状可在一个个体上交叉吗?
不同性状可在同一个个体上交叉,如以株高和颜色性状划分群体,两个群体间会有重叠的个体存在,不影响分析的结果。
Q6:自然群体GWAS的一般研究策略有哪些?
(1) 复杂性状定位到单基因水平,获得目标性状相关的功能标记;
(2) GWAS和群体进化结合,对控制目标性状的候选基因进行精准定位;
(3) GWAS和QTL互补定位目标性状的候选基因。
Q7:客户提供代谢组数据,是否可以用来做关联分析?
可以,基因表达 GWAS(eGWAS)、代谢组 GWAS(mGWAS)、蛋白组 GWAS (pGWAS )等,是从表型的角度对 GWAS 方法进行的优化,通过将从基因到生态表型过程中的基因表达,代谢物作为分子表型与SNP等基因型进行关联,这种方法和常规表型的GWAS分析方法原理、软件模型都一样。
更多相关问题可咨询派森诺,全程专业服务!
派森诺动植物领域项目经验丰富




















