

2025-01-14

一、微生物多样性常见问题
1.微生物组常用的产品有哪些?
答:微生物组常用的产品包括微生物多样性,宏基因组和宏转录组等。
微生物多样性也叫扩增子测序,是一种广泛应用于环境微生物群落结构研究的技术。它主要通过PCR(聚合酶链反应)技术对特定的基因片段进行扩增,然后通过对这些扩增产物进行高通量测序来分析样品中微生物群落的组成和多样性。
宏基因组(Metagenome)和宏转录组(Metatranscriptome)是通过鸟枪法测序技术(Shotgun sequencing),结合全微生物组关联分析(Microbiome-Wide Association Studies,MWAS)的策略,分别从DNA/RNA水平,全面精细地展示整个微生物群落的物种组成谱、功能代谢谱、表达谱,进而从原理上阐明微生物群落在生态系统中发挥作用的根本机制。
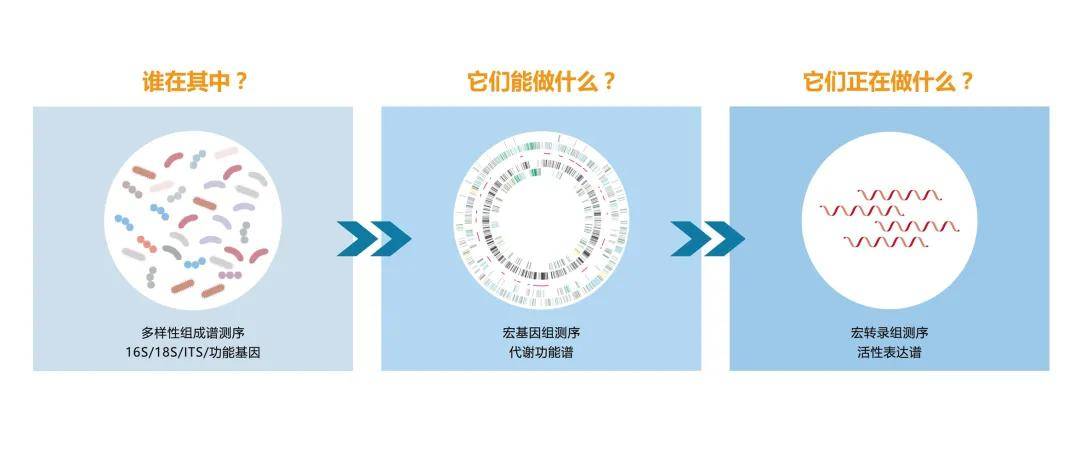
多样性组成谱测序主要研究微生物群落的物种组成、物种间的进化关系以及群落的多样性。
宏基因组/宏转录组测序是对群落中所有微生物的基因序列或其对应的转录本进行研究,因此,不仅可以获得编码这些基因的物种信息(可以深入到种甚至菌株水平)还可以对测到的众多基因进行功能层面的更广泛的研究。
因此,多样性组成谱关注微生物物种多样性、组成和它们的丰度分布,宏基因组除了可以得到微生物群落的物种组成的精细信息外,还可以了解群落中微生物所携带的基因功能特性,及其所涉及的功能通路,而宏转录组则可以看作是宏基因组的活性表达的“子集”,是对群落中具有活性的微生物、及其正在表达转录的基因功能进行研究。简而言之,多样性组成谱测序能够告诉我们“谁在其中”,宏基因组测序则能展现群落中各微生物成员的“分工职责”,而宏转录组测序将揭示这些微生物“正在做哪些事情”
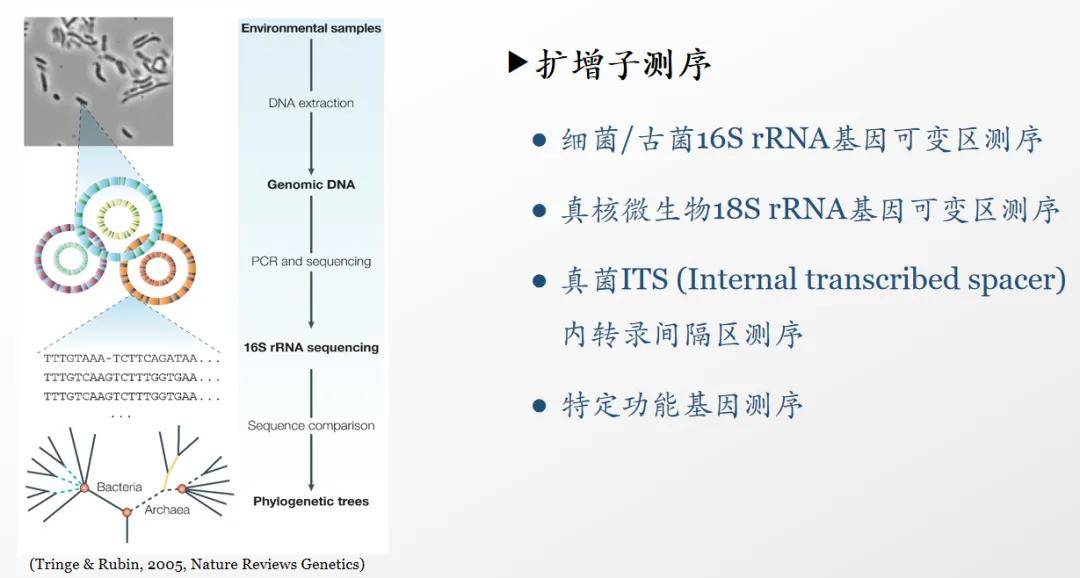
2. 菌群多样性组成谱测序,该如何选择 rRNA 基因的可变区 (V区)?
答:1)对于原核微生物群落(细菌或古菌)而言,16SrRNA基因的9个可变区从理论上来说都是可以选择的,但根据我们大量的实验以及发表文献统计结果,结合各V区的长度,目前,二代Illumina 测序中应用比较多的单V区是V4区(~250bp),双V区是V3-V4区(~470bp)或V4-V5区(~420bp)。另外,如果是对 16S rRNA 基因进行三代 PacBio 测序,则能够覆盖整个 16S rRNA 基因全长(大约1.6 kb);
2)对于真菌菌群而言,相比18S rRNA 基因,ITS序列的进化速率更快,能够累积更多的物种变异,因此对于真菌物种检测的分辨率更高。目前,二代Illumina 测序中应用最多的是ITSI区,ITS2 区次之,而三代 PacBio 测序则可以覆盖ITS 全长,即ITS1 区 +5.8S rRNA 基因 +ITS2 区;
3)除真菌外,对于其它真核微生物而言,二代 Illumina测序中应用最多的是 18SrRNA 基因的 V4 区,而三代 PacBio 测序则可以覆盖 18S rRNA 基因的近全长。
 扩增子实验流程以及扩增区域选择
扩增子实验流程以及扩增区域选择
3.微生物多样性同批次测序的各个样本,测序得到的序列数有差异,对数据分析会有影响吗?
答:同批次测序的各个样本,测序得到的序列数不可能一模一样,总会存在一定程度的波动,这是正常现象。序列数的差异与初始样本量以及文库定量等环节都有一定关系。一般来讲,由于这些样品是一起建库测序的,序列数的变化范围比较有限。另外,我们在分析过程中,会对每个样本测到的序列数进行抽平处理,从而消除由于测序量的差异导致的菌群组成结构上的不一致。
4.对于多样性组成谱测序、有哪些常用物种注释数据库?各有哪些特点?适用于哪些微生物类群?
答:常用的物种注释数据库包括 Greengenes、SILVA、NCBI、UNITE、RDP、Greengenes2等数据库。
1)Greengenes 数据库 13.8版本收录了1,262,986条16S rRNA序列,是专门针对细菌、古菌 16S rRNA 基因的数据库。该数据库注释偏向于样本为人源、肠道类样本,注释效果较好,物种命名也比较规范,只是数据库版本比较老,2013年8月之后没再更新过;
2)SILVA 数据库包含细菌、古菌和真核生物的分类学信息。原核生物的 16S 和真核生物 18S rRNA 基因都可以用该数据库注释分析。该数据库注释偏向于环境方面的样本,如土壤、水体、空气等样本,当然如人源与肠道方面的样本也可用该数据库注释,用途比较广泛。但是,该数据库的物种命名,不如Greengenes 规范;
3)NCBI数据库包含提交NCBI序列数据库相关的所有生物体的名称,并且每日更新。可用于细菌、古菌、真菌和病毒序列的注释分析,一般用于功能基因方面的注释及真核 18S rRNA 基因数据注释;
4)UNITE 数据库包含的是高质量 ITS参考序列,是专门针对真菌ITS序列的数据库;
5)RDP数据库基于来自细菌、古菌和真菌的rRNA 基因序列。可用于细菌和古菌16S rRNA 基因和真菌 28S rRNA 基因的注释分析,物种命名比较规范,但 RDP 数据库注释只能到属水平,且 2016年9月30日之后就没再更新过。
此外,对于某些特定的微生物类群,我们也能选择针对性的数据库进行物种注释。比如,MaarjAM 数据库可用于 AMF 丛枝菌根真菌群落的注释,HOMD 数据库可用于人体口腔菌群的注释,PR2 可用于原生生物群落的注释。
6)Greengenes2 数据库:2023年7月,Nature Biotechnology(IF: 46.9)发表了Daniel McDonald(通讯作者Rob Knight)等人的文章,引入了Greengenes2。这是一个参考树,通过将序列插入到全基因组系统发育树中,将基因组和16S rRNA数据库统一在一个一致的、集成的资源中。结果表明从相同的样本中生成的16S rRNA和宏基因组数据在主坐标空间、分类学和表型效应大小上是一致的。此外,Greengenes2数据库的系统发育覆盖率远大于SILVA、Greengenes和GTDB等数据库。Greengenes2数据库将会是微生物组研究的一个利器。

微生物多样性物种注释数据库
更多微生物组数据库详细介绍请查看 往期软文
扩增子和宏基因组测序常用数据库介绍
5.多样性组成谱测序,可以鉴定到种吗?
答:在以16S/18S rRNA基因或ITS区域为目标序列的菌群多样性组成谱测序研究中,Illumina等二代测序平台一般只能对单个或连续的两三个可变区序列进行测序分析,通常情况下无法获得 rRNA 基因全长序列的信息,因而往往只能在属水平进行物种鉴定分析,而难以在种等精细水平识别测得的序列,难以获取“高分辨率”的微生物物种信息,菌群中大量成员对应的物种信息仍处于“模糊”状态。
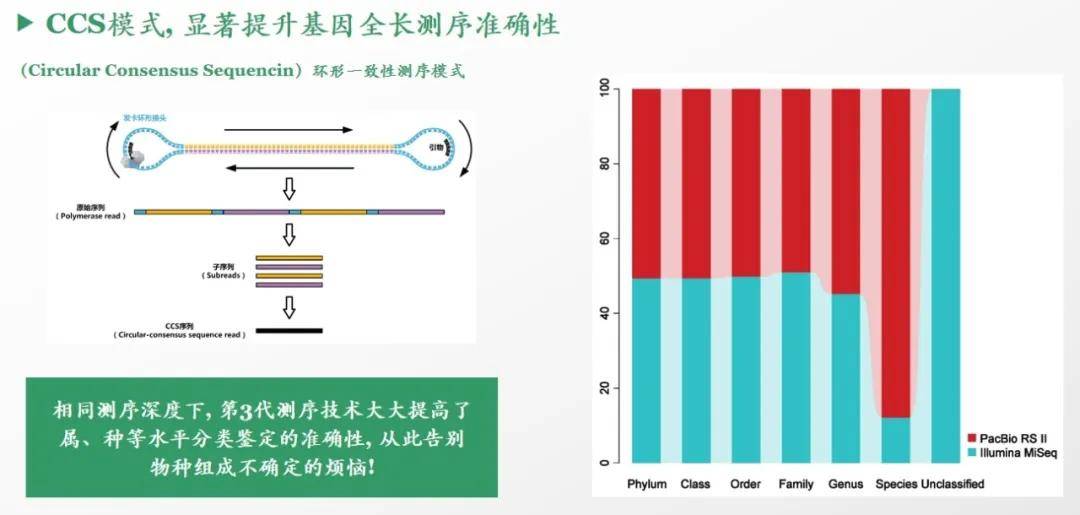
而以 PacBio 公司的 Sequel测序系统为代表的三代测序技术,利用专利的 SMRT单分子实时测序技术,可以对DNA序列实现单分子级别的超长读长测序,因而能够轻易读取微生物的 rRNA 基因全长序列;其次,通过PacBio所独有的环形一致性测序模式(Circular-consensus sequence,CCs),可以极大地提高单基测序的准确率,般而言,将 rRNA 基因全长序列循环测序5~6次后,获得的一致性序列的准确率可以达到 99.99%(QV40)以上,远远超过了二代测序的准确率,从而充分保障获得的rRNA基因全长序列的精确性。
因此,在相同测序深度下,PacBio的rRNA基因全长测序在种水平的分辨能力大大超过了 Illumina 的短片段测序,使菌群组成得到精准解析;不仅如此,由于读长较短Illumina 会产生“无法确定归属(Unclassifed)”的物种,但这些物种的分类学信息都能通过 PacBio 全长测序得到准确识别。因此,PacBio 的SMRT 测序技术能够大大提高我们在种水平解析菌群精细组成结构的能力。
当然,这也并非意味着Illumina测序技术不适合多样性组成谱测序,只是无法在种等精细水平,获得物种组成的全面信息。因此,如果进行Illumina 测序,我们建议是在属水平进行相应分析,但如果有特殊需求,需要在种水平进行分析,我们也可以开展,只是可能会出现大部分物种无法注释到种水平的情况(也比较容易引起审稿人质疑,因为片段较短);而如果是进行 PacBio 测序,我们默认都可进行种水平的分析。
 三代微生物多样性
三代微生物多样性
6.多样性组成谱测序能分析功能基因吗?功能基因测序和多样性组成谱测序,有什么差别?
答:多样性组成谱测序主要是基于rRNA 基因的可变区(如16SrRNA 基因某V区或真菌 rRNA 基因的 ITS 区域)进行环境微生物群落的多样性和组成丰度的鉴定,因此并不是直接基于某个功能基因的片段进行的测序和分析。但是我们有一项特色分析内容“菌群代谢功能预测”,可以根据测到的物种,推断它们可能具备的功能特性。
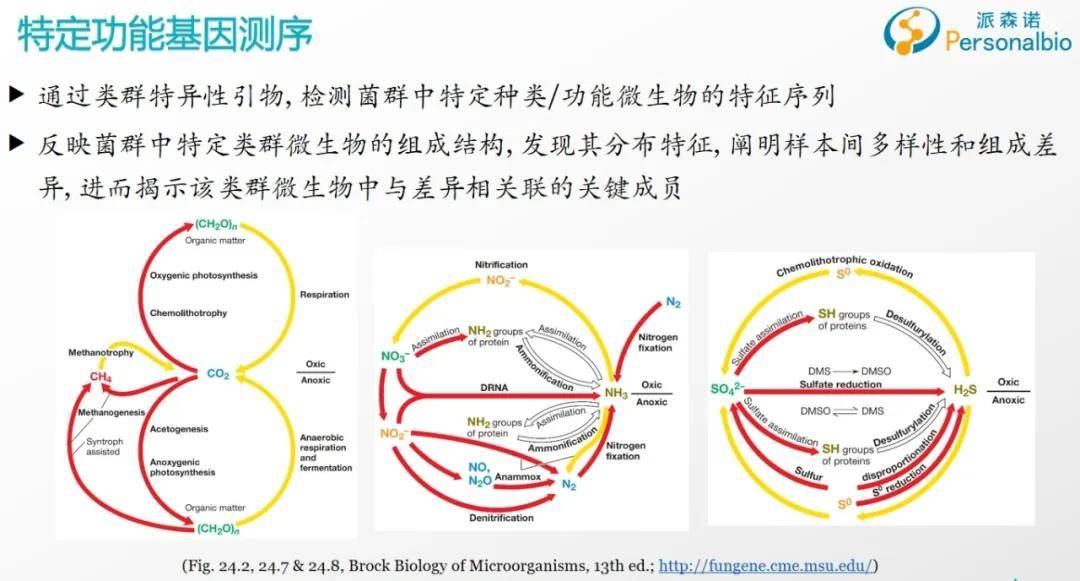
不过,如果老师关注的是某一个功能基因的研究,我们建议您可以直接对这一功能基因开展测序,而不需要测定rRNA 基因的多样性组成谱,因为rRNA 基因测序获得的是广谱的微生物类群,而功能基因测序则可以避免测出与关注功能不相关的微生物物种。另外,如果老师同时关注整个群落中的多个基因,我们建议您可以考虑采用宏基因组或者宏转录组测序,这样可以获得更为全面的微生物编码的功能信息。
功能基因测序是扩增某一功能的标志性基因序列,解析具有这一功能的微生物组成谱,需要用相应的特定基因引物进行该基因的PCR扩增,然后进行测序,反映的是该特定功能类群微生物的组成结构,一般采用NCBI数据库对功能基因的测序数据进行物种注释,常见的功能基因包括氮代谢的基因,比如amoA、nirS、nirK、nxrA 等,以及碳代谢和磷代谢相关的功能基因;而多样性组成谱测序多是细菌/古菌16SrRNA基因/真菌 ITS 内转录间隔区进行测序,反映的是细菌/古菌或真菌的广谱种群的组成结构,可以通过 PICRUSt 等方法预测菌群整体所具有的潜在功能特性,关注的对象并不局限于执行某一类特定功能的微生物。
 功能基因测序
功能基因测序
二、宏基因组与宏转录组常见问题
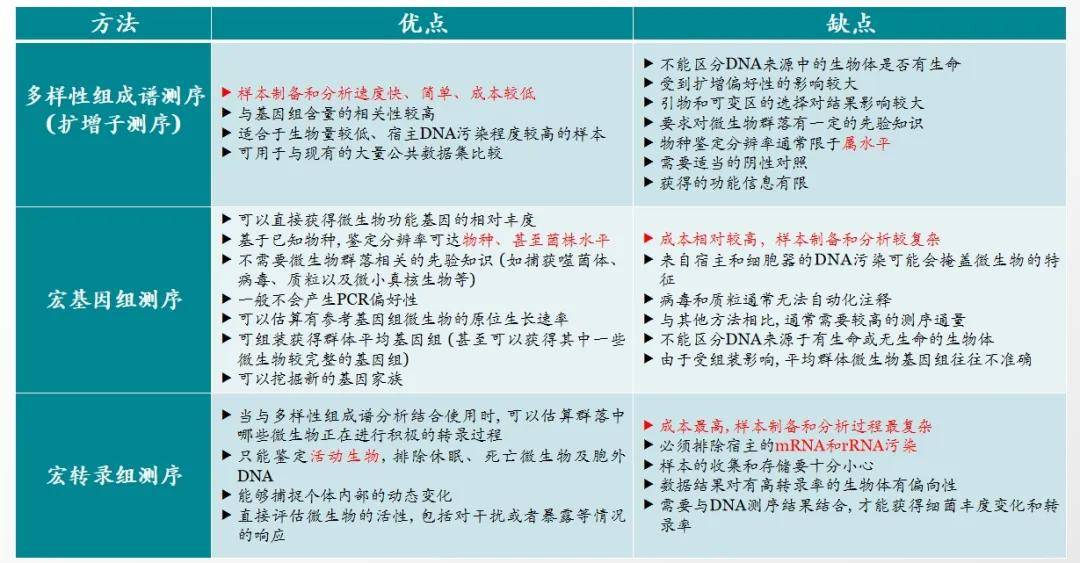
1.宏基因组与微生物多样性和宏转录组的主要区别是什么?我设计实验的时候该如何选择?
答:微生物多样性与宏基因组和宏转录组的区别如下
2.人体或动物的共生菌群进行宏基因组/宏转录组测序,会不会有宿主序列的污染?
答:会受到宿主序列污染的,但污染程度根据实际情况,可能或大或小。为尽量避免宿主的序列干扰,
我们建议从两个方面进行优化:
1)取样过程中,尽量避免取到宿主的组织;
2)适当提升测序数据量,测序完成后,可以将测序数据与宿主的参考基因组进行比对,过滤来源于宿主的序列,将过滤后的数据应用于后续分析。
3.为什么宏基因组与多样性组成谱的物种注释结果存在差异?
答:无论是宏基因组,还是多样性组成谱,都会分析群落样本中物种的种类组成和分布结构,但是物种的丰度组成未必一模一样。一般情况下,两种方法获得的优势物种种类应该差别不大,但是丰度上可能会可能存在少许差异。
究其原因,一方面是因为多样性组成谱是基于核糖体小亚基基因序列进行物种注释和丰度统计,但是在不同物种中核糖体基因的拷贝数不一样,这会带来一些误差;另一方面,多样性组成谱需要对群落 DNA 进行 PCR扩增,一般是在属水平进行物种组成谱的解析,而宏基因组是直接对基因片段进行鸟枪法测序,然后通过拼接组装来预测基因信息,进而获得物种组成谱,可以深入到种水平,两者的方法策略是截然不同的,因此也会对物种的丰度组成产生一定影响。其次,两种方法的测序深度是截然不同的,一般多样性组成谱的测序量在 3~6万条序列/样本的水平,而宏基因组一般是在 5~10G的测序数据量,因此测序深度也会导致检测到的物种种类有所不同。
4.宏基因组/宏转录组测序,如何将功能和物种两方面的信息对应起来?
答:宏基因组/宏转录组测序项目,会根据测序数据,进行序列拼接,并将拼接到的 Contig/Scaffold 序列进行基因预测,然后对这些基因,同时进行功能注释和物种注释。也就是说,无论是宏基因组,还是宏转录组项目,只要获得了基因信息,就可以同时掌握该基因编码了什么功能,又来源于哪些物种,即:可以根据基因信息,将微生物的物种组成及其携带的功能通路信息对应起来。这里小派提醒大家如果在派森诺做的宏基因组和宏转录组测序项目可以使用派森诺基因云个性化创建数据集将物种和功能对应起来哦!
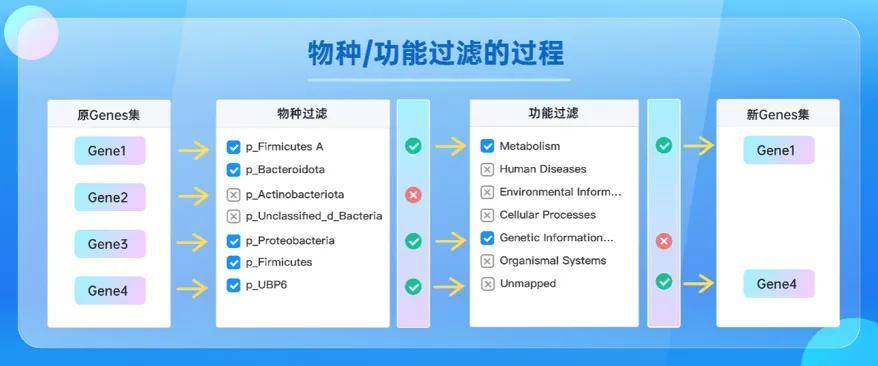 派森诺基因云数据集创建流程
派森诺基因云数据集创建流程
5.宏基因组/宏转录组测序,常用的数据库有哪些?
答:农学和医学方向对宏基因组/宏转录组数据库的应用有所不同。农学常用的数据库包括MCycDB,NCycDB,PCycDB,SCycDB,CAZy,BacMet, PlasticDB等;
医学常用的数据库包括PHI,VFDB,MicroPhenoDB等,同时也有一些农学和医学方向通用的数据库KEGG,eggNOG,GO,Swiss-Prot,Pfam,Metacyc等。派森诺宏基因组与宏转录基因云全新升级包含30+数据库同时满足农学和医学方向数据分析的需求
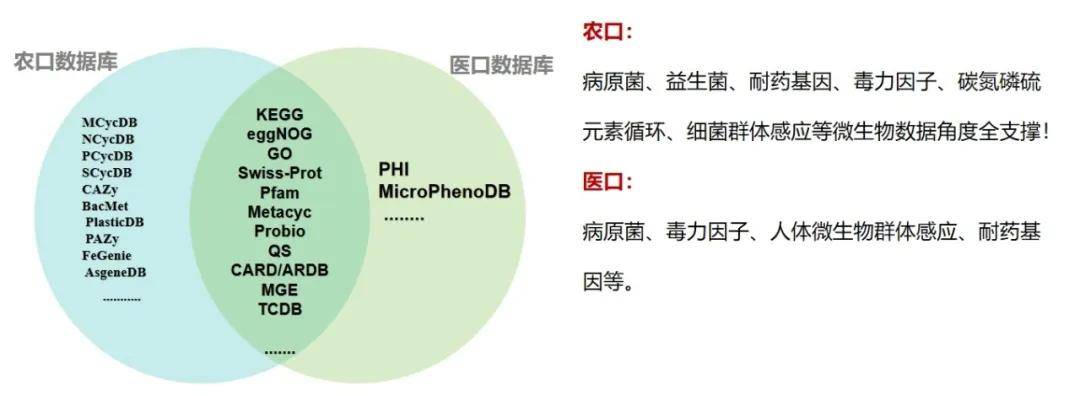 派森诺宏基因组与宏转录组30+数据库
派森诺宏基因组与宏转录组30+数据库
三、其它常见问题
1.微生物组高通量测序,较基因芯片检测的优势在哪?
答:一般而言,基因芯片能检测的,高通量测序都能做;但反过来,高通量测序能检测到的,基因芯片不一定能做。这是因为,基因芯片是通过与一组已知序列的核酸探针杂交进行定量检测,因此无法对未知的物种和基因进行检测,且杂交检测的方法会存在较高的假阳性;而高通量测序方法则能轻松解决芯片技术在检测小分子时遇到的短序列、高度同源等技术难题,同时测序方法还能检测到未知的物种和基因片段信息。随着高通量测序的通量越来越大、成本越来越低,测序技术在微生物组研究领域的应用,也越来越广泛,已成为微生物组研究的常规检测手段。
2.微生物组测序完成后是否需要验证?可以用 qPCR 方法验证吗?
答:可以通过荧光定量PCR(GPCR)进行验证。一般而言,qPCR获得的是微生物的绝对丰度,而测序技术一般获得的是群落中不同物种/基因/转录本的相对丰度百分比,因此,这两种方法并不完全一致,但可以相互印证。
如果是进行多样性组成谱或者功能基因测序,可以针对总菌数或者某一类特定的微生物类群的绝对丰度设计特异性引物进行验证;而对于宏基因组/宏转录组而言,则可以针对某一类功能基因或对应的转录本,设计引物验证其基因拷贝数或转录本表达量的变化。
3.微生物组测序是否需要生物学重复?重复几次?
答:需要。一般而言,微生物群落样本总是存在一定的随机性和不均一性,而设置生物学重复将有助于评估样本间差异程度,同时增强结果的可靠性。测序的样本数越多,越能够降低组内样本差异而引入的影响。此外,足够数量的生物学重复,还有助于检测离群样本,因为异常样本的存在,会严重影响检测结果的准确性。在生物学重复数量足够的情况下,通过分析菌群组成、Alpha和 Beta 多样性指数,可以发现异常样本,将其剔除。
如果样本的类型是动植物样本,如肠道内容物或粪便样本、组织样本等,个体间差异较大,建议 10个以上生物学重复;如果样本的类型是环境样本,如土壤、水体、空气等,建议6个以上生物学重复;提高生物学重复的数量可以大大减小生物个体间存在的误差。
4.微生物组样本的采集和运输有什么要求?
答:不同样本的微生物组样本的采集是不同的,部分样本的采集需要特殊的方法。农学和医学具体的样本的采集方法可以咨询派森诺当地的销售小伙伴获取。


派森诺微生物组农学和医学取样方法


















