

2025-03-27

Highlights
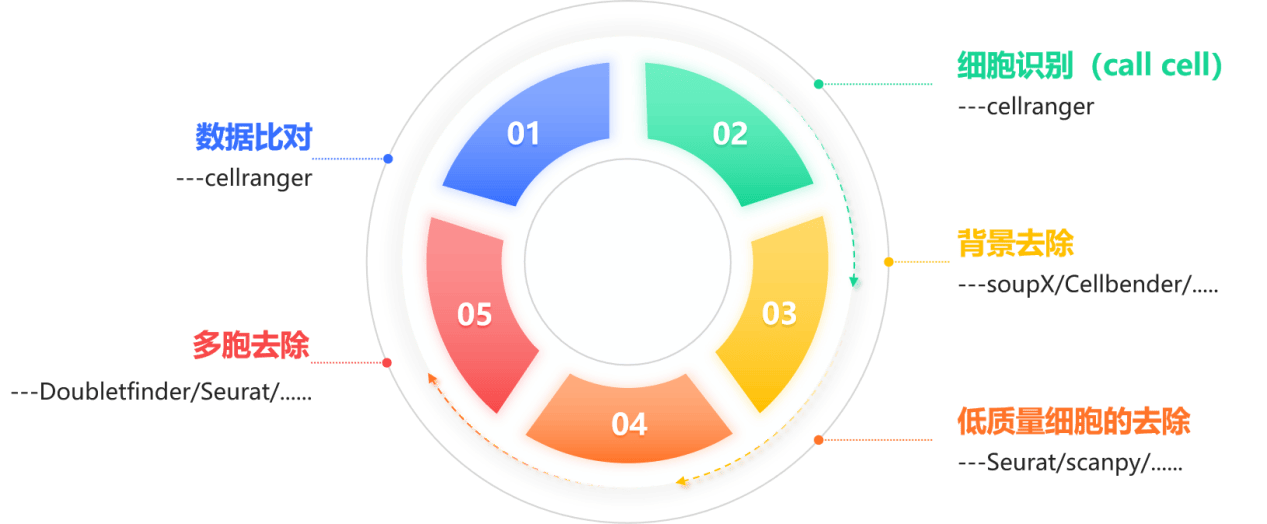
1.本文系统梳理单细胞转录组测序关键质控流程,涵盖数据比对、细胞识别、背景去除、低质量细胞去除、多胞去除等环节,详细解析 Cell Ranger、soupX、Cellbender、Seurat、Scanpy、DoubletFinder 等主流软件技术原理。
2.单细胞转录组测序质控流程可解决实验数据失真问题,通过各环节质控与软件协作,为后续细胞类型分析、状态研究及细胞间相互作用探究提供可靠数据支撑。
3.派森诺单细胞转录组依托 10x Genomics 平台,优化质控流程,提供样本处理至数据挖掘标准化服务,确保数据的高质量与可用性,助力老师科研。
单细胞转录组测序(scRNA-seq)技术通过解析单个细胞的基因表达图谱,为生命科学研究开辟了全新维度。然而,实验过程中不可避免的空液滴、低质量细胞、双胞污染及环境 RNA 干扰,可能导致数据失真。本文系统梳理单细胞转录组测序的关键质控流程,揭示主流软件的技术原理与应用价值。
一、数据比对——Cell Ranger的精准定位术
数据比对是将测序得到的原始读段(cDNA片段)与参考基因组或转录组进行匹配,确定其在基因组上的位置,为后续分析提供关键支撑。Cell Ranger 作为 10x Genomics 平台的配套工具可以通过以下流程,高效地将复杂的测序数据转化为直观的基因表达矩阵。

流程如下:
1.样本拆分:通过cellranger mkfastq将混样测序数据按样本标签拆分为独立的fastq文件。
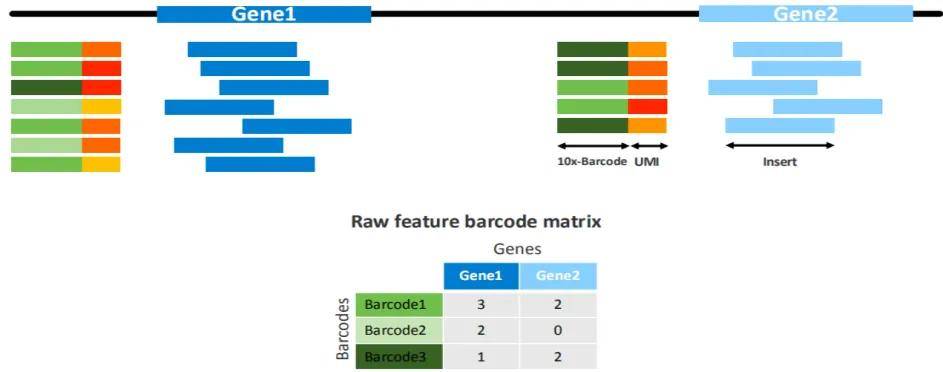
2.序列提取:从测序数据的 R1 端提取 16bp 的细胞条形码(Barcode)和 12bp 的 UMI 序列,Barcode 用于区分不同细胞,UMI 则能精准计数基因表达量,而 R2 端数据主要用于基因比对。
3.参考基因组比对:基于STAR算法,将测序片段比对到参考基因组,并区分外显子、内含子和基因间区(规则:50%以上比对到外显子才记为外显子区)。
4.质量控制:校正Barcode(允许1个碱基错配),过滤无效UMI(如含AAAAAAAAAA的同聚物),最终生成原始表达矩阵,为后续分析奠定基础。
二、细胞识别:Cell Ranger 精准定位 “真细胞”
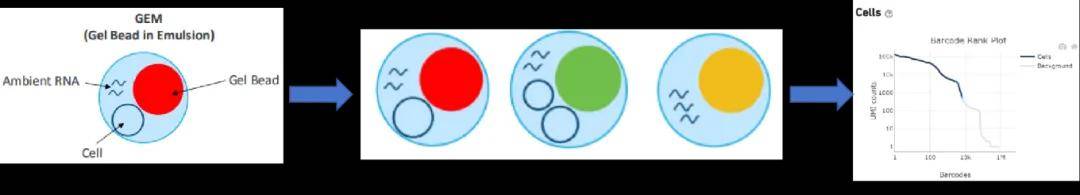
在单细胞测序实验中,每个细胞会被分配唯一的细胞条形码,Cell Ranger 会识别这些条形码。它通过统计每个条形码关联的测序 reads 数和独特分子标识符(UMI)数,评估 “潜在细胞” 的测序深度和数据质量。高质量细胞数据具有充足测序深度和合理 UMI 计数,利于后续细胞精准识别。
Cell Ranger 软件主要根据 UMI 计数分布识别细胞,通过每个细胞条形码对应的 UMI 计数构建分布。通常,真实细胞的 UMI 计数远高于背景噪音,软件通过设定基因表达量阈值识别“潜在细胞”,“潜在细胞” 的表达基因数量或总体表达量高于阈值则被认定为真实细胞,反之则为背景噪音。背景噪音的基因表达量低且分布均匀,与真实细胞差异明显。
某些情况下,自动识别可能无法准确判定细胞数量。如样本细胞活性差、捕获效率低或存在大量背景 RNA 污染时,自动识别的细胞数量可能低于实际。此时可借助条形码秩图,利用 force - cells 功能重新识别被误判为背景的细胞并纳入统计,使最终细胞数更符合实验预期,确保后续数据分析可靠。
三、背景去除:soupX 与 Cellbender “净化” 数据
在实际的单细胞测序数据常受到背景信号的干扰,这些背景信号来源于细胞外 RNA 污染、细胞破碎后的 RNA 泄露或样本处理过程中的杂质。背景信号会导致虚假基因表达信息,模糊细胞间的基因表达差异,影响对细胞类型、状态和相互作用的分析。此时需借助软件去除,主要有两种soupX和Cellbender。
注:单细胞测序实验中通过 poly (A) 捕获已自然排除核糖体 RNA 等背景,且质控聚焦于过滤低质量细胞(如线粒体基因比例、检测基因数阈值)已经做了背景去除,所以一般正常情况下为了保留更多的低丰度转录本的异质性信号无需背景去除。
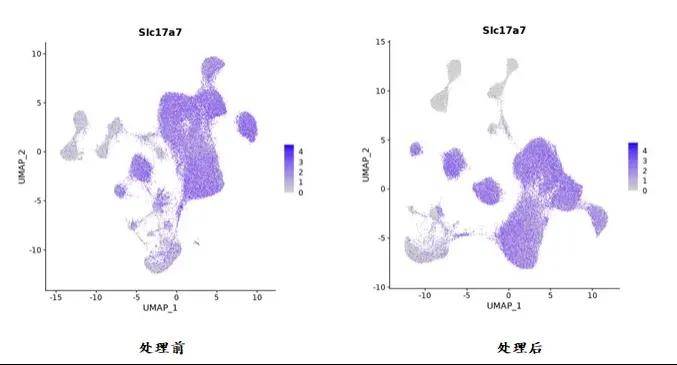
SoupX 专注于去除因细胞裂解导致的游离 RNA 污染。它通过统计分析,依据基因在不同细胞中的表达模式,识别出在背景高表达、细胞内低表达的基因,估算背景组成并从原始基因表达矩阵中减去,使数据更真实地反映目标细胞的表达情况。下面左图中Slc17a7是小鼠的兴奋性神经元经典marker,在某些非神经元细胞中也高表达,是比较典型的环境RNA污染,去除 Slc17a7 等环境 RNA 污染后,神经元 marker 表达更清晰(右图)。
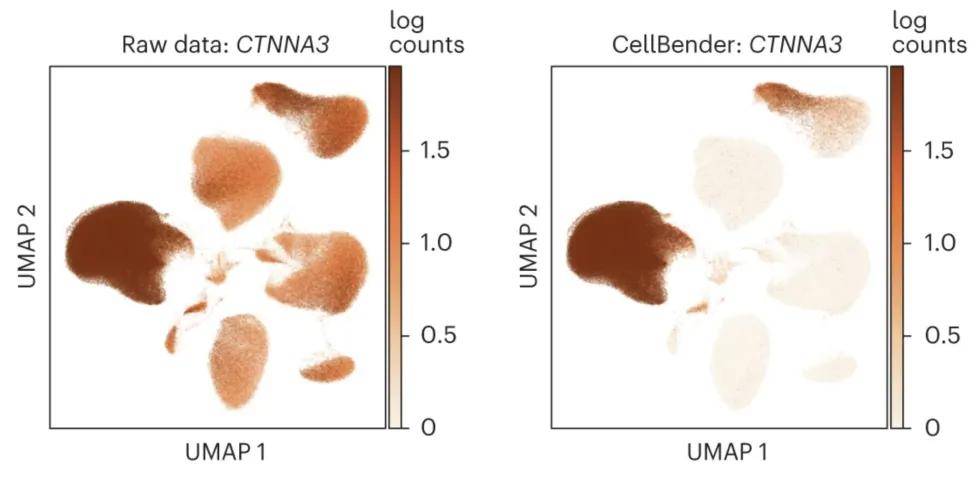
Cellbender 是基于深度学习的软件工具,应用更为广泛,它能处理多种复杂背景污染,包括游离 RNA、空液滴、低质量细胞和环境 RNA 等。Cellbender 将背景噪声分解为液滴外 RNA 渗入和液滴内自由 RNA,利用分子捕获概率模型区分真实信号与噪声。下图基因 CTNNA3,这是一种参与细胞间粘附的蛋白质编码基因,在 CellBender 之前(左)和之后(右)具有已知和特异性的表达模式。作为参考,CellBender 加工后 CTNNA3 高表达的两个细胞簇是心肌细胞(左中簇)和血管平滑肌细胞(右上簇)。
四、低质量细胞去除:Seurat 与 Scanpy “剔除瑕疵”
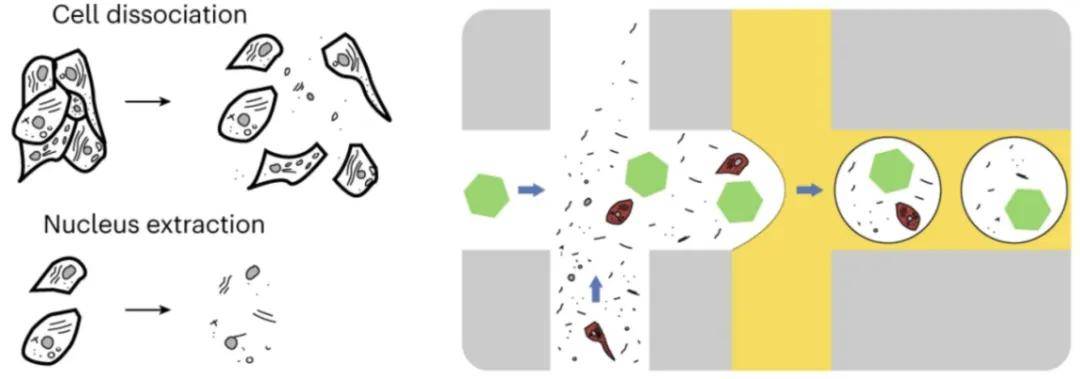
单细胞测序在上机前需要对样本进行解离,由于解离需要用到大量消化细胞间基质的酶,这些酶或多或少会对细胞的状态产生影响,有的可能会导致细胞发生应激,有的甚至会直接导致细胞膜破碎并死亡,由此便产生了一些低质量的细胞,下游数据表现为UMI计数偏低、基因检出数较少、线粒体基因占比异常升高以及核糖体基因比例偏离正常范围等特征。这类细胞受实验操作、细胞自身状态等因素影响,数据质量欠佳,会干扰后续分析结果。此时需借助软件去除,主要有两种Seurat和Scanpy。
一般常用的是Seurat,主要原因是Seurat(R语言)通过计算每个细胞的 UMI 计数、检测到的基因数量、线粒体基因比例等指标来对数据进行质量控制,识别并去除低质量细胞。且Seurat提供标准化质控函数,函数经 10x Genomics官网大量标准数据集验证,阈值参数直接参考 CNS 文章,且90% 以上单细胞领域 CNS 论文采用 Seurat 质控标准,确保分析结果的可重复性与方法学权威性。
Scanpy(Python)专为大规模数据设计,支持百万级细胞处理。其运用内置函数计算细胞各项质量指标,包括细胞总计数,即每个细胞中所有基因表达量总和检测到的基因数以及线粒体基因比例,经加载数据、算指标、设条件、筛选细胞等流程去除低质量细胞。
✮重要参数
(1)nFeature_RNA:过滤掉总的基因数大于7000或者小于400的细胞,过高的基因数可能为双细胞或多细胞,过少的基因数可能是空液滴或低质量细胞;
(2)nCount_RNA: 过滤掉总的UMI数大于50000的细胞,单个细胞的总 RNA 量有限,过高 UMI 可能是由于实验过程中两个细胞进入了一个微滴,过低可能因细胞过小(如血小板)或捕获效率低导致数据不可靠,这类数据需要去除。
(3)percent.mito:过滤掉线粒体基因表达占比大于20%的细胞,正常细胞中,线粒体基因比例很低,除了一些特殊的代谢旺盛的组织类型如肾脏组织,凋亡中的细胞通常线粒体基因表达异常高,线粒体基因表达量可以作为鉴定样本中低质量细胞的参照指标。抽核样本此参数一般卡到5%以下,因线粒体 DNA 主要存在于细胞质。
五、多胞去除:DoubletFinder 联合 Seurat “拨乱反正”
scRNAseq的理想情况是每个barcode 下只有一个细胞,但在实际情况中会有两个或多个细胞共用一个barcode,称之为 doublets。这些细胞主要特点是检测的UMI数和基因数往往比正常细胞要多一倍及以上,另外可能会带有不同细胞类型的经典marker基因。多细胞会混淆不同细胞的基因表达,增加数据中的噪音。因此,去除结果中的多细胞可以提高数据的准确性,有助于后续提供更精确的生物学解释。
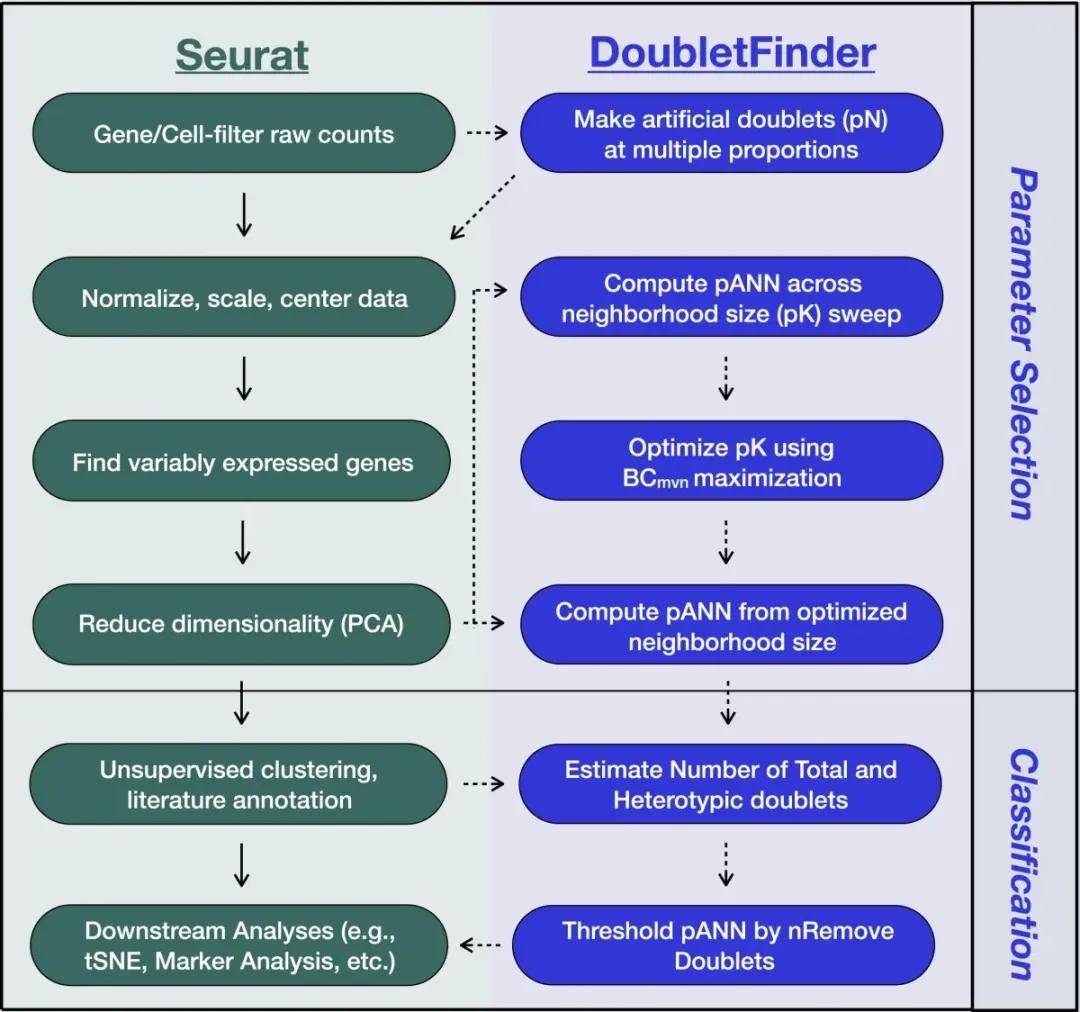
常见多胞去除方法是Seurat 联合DoubletFinder共同协作进行。Seurat 可根据单个细胞中的 max gene 数初步判断 doublet,DoubletFinder 是一款 R 语言包,它从现有矩阵的细胞中模拟一些双细胞,计算每个细胞与模拟双胞的相似性,相似性越高,该细胞为真实双胞的可能性就越大。二者联合通过模拟生成 doublets、计算每个细胞的最近邻、计算最近邻中的模拟 doublets 数量得到 pANN 排序,再根据期望 doublets 数量设置 pANN 阈值,从而过滤掉 doublets。
六、总 结
单细胞质控软件通过多维度协作,构建了从数据比对到多胞去除的完整质控体系。随着算法优化与硬件升级,质控流程将向自动化、智能化方向持续演进,为单细胞研究提供更可靠的数据保障。


















