2025-03-29

目录
1.IGV软件安装和基本界面
2.转录组reads覆盖查看
3 表观组学peak峰图查看
1、IGV软件安装和基本界面
IGV(Integrative Genomics Viewer)是一款本地即可使用的可视化工具,安装简单,操作便捷,支持多种格式的转录组、表观组等数据的可视化分析!只需导入参考基因组文件以及bam/bw文件(IGV支持多种文件类型,这里我们主要以结果提供给老师的bam/bw文件为例),即可对转录组、表观组的比对结果进行可视化浏览!
IGV官网:https://igv.org/doc/desktop/
IGV下载:https://igv.org/doc/desktop/#DownloadPage/
备注:IGV软件需要在安装java的前提下打开,因此可以直接下载Java included的版本。
也可以通过网盘下载:https://pan.baidu.com/s/1baZ9MF9wEVi4_eqhgl82zg?pwd=m5i7
基本界面:
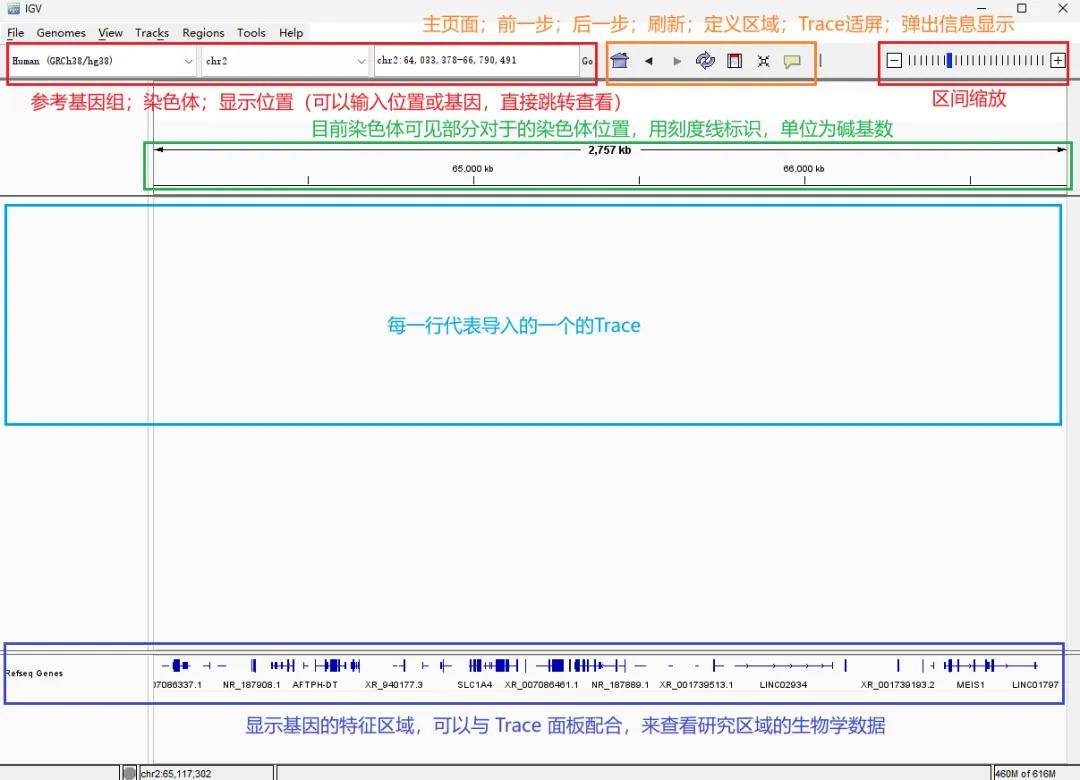
重要菜单栏(File、Genome、Tools):
File → Load from File → 加载文件(加载基因注释gtf/gff文件、样品bam/bw文件等);
File → Save PNG/SVG Image → 导出PNG/SVG格式的图片;
Genome → Load Genome from File → 加载基因组文件;
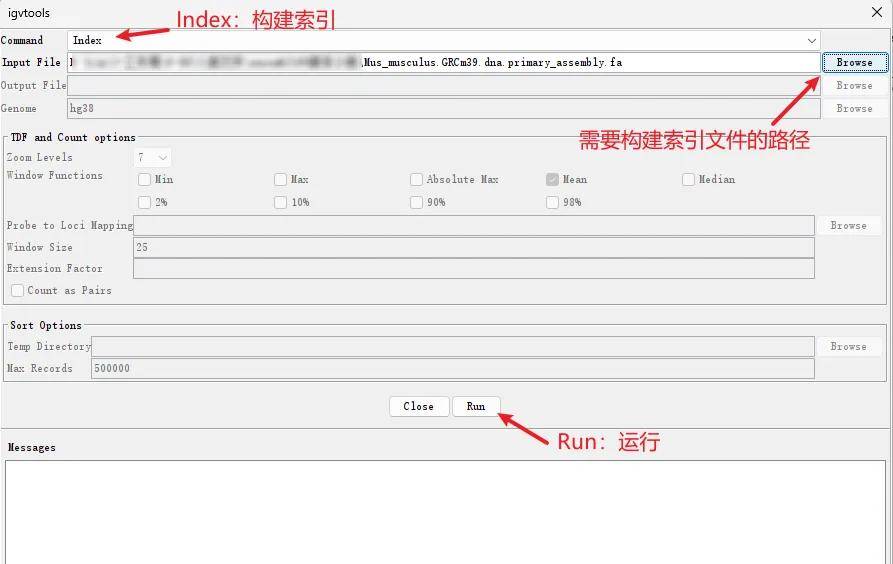
Tools → Run igvtools → IGV生成索引的重要工具
选中Index工具,可以生成fasta文件的索引fai文件,可以生成bam文件的索引bai文件。
备注:部分原核的bam文件可能需要先选sort工具对bam文件进行排序,再对得到的sorted bam文件使用Index构建bai索引。
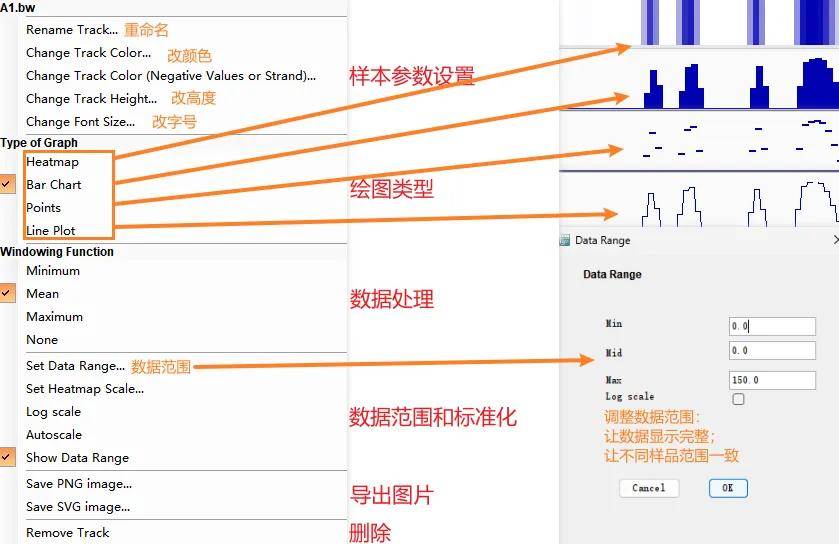
在Trace窗口可以右键呼出如下菜单栏,进行调整(修改颜色、数据范围等)。
在基因组注释窗口可以右键呼出如下菜单栏,选择基因展示形式(可以展开多条转录本)。
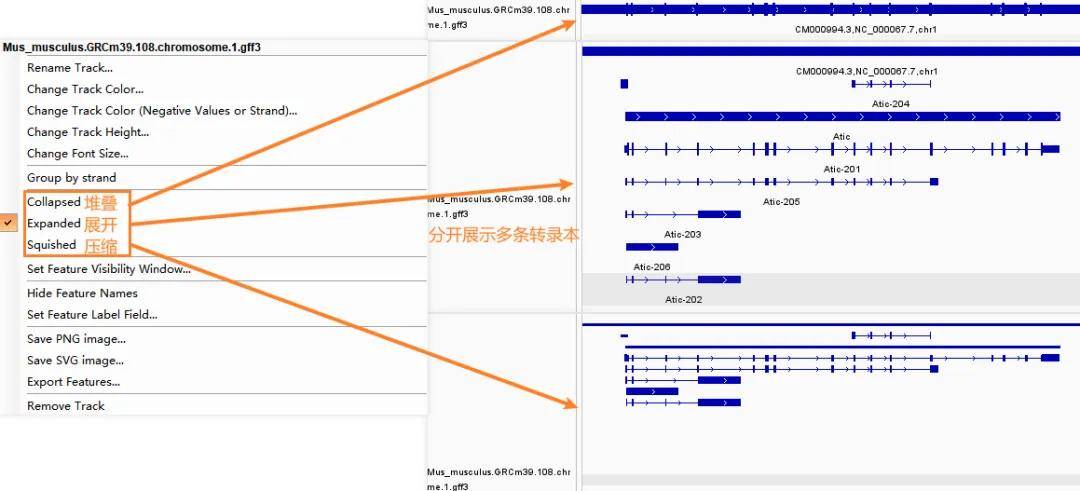
2、转录组reads覆盖查看
a.文件准备
参考基因组的序列文件和结构注释gtf/gff文件、样品对应的bam/bw文件。
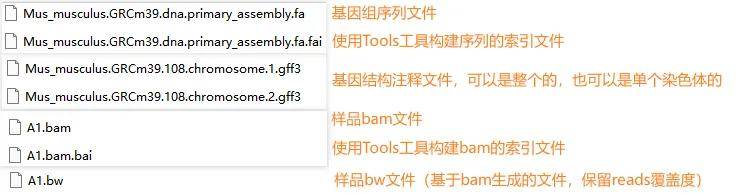
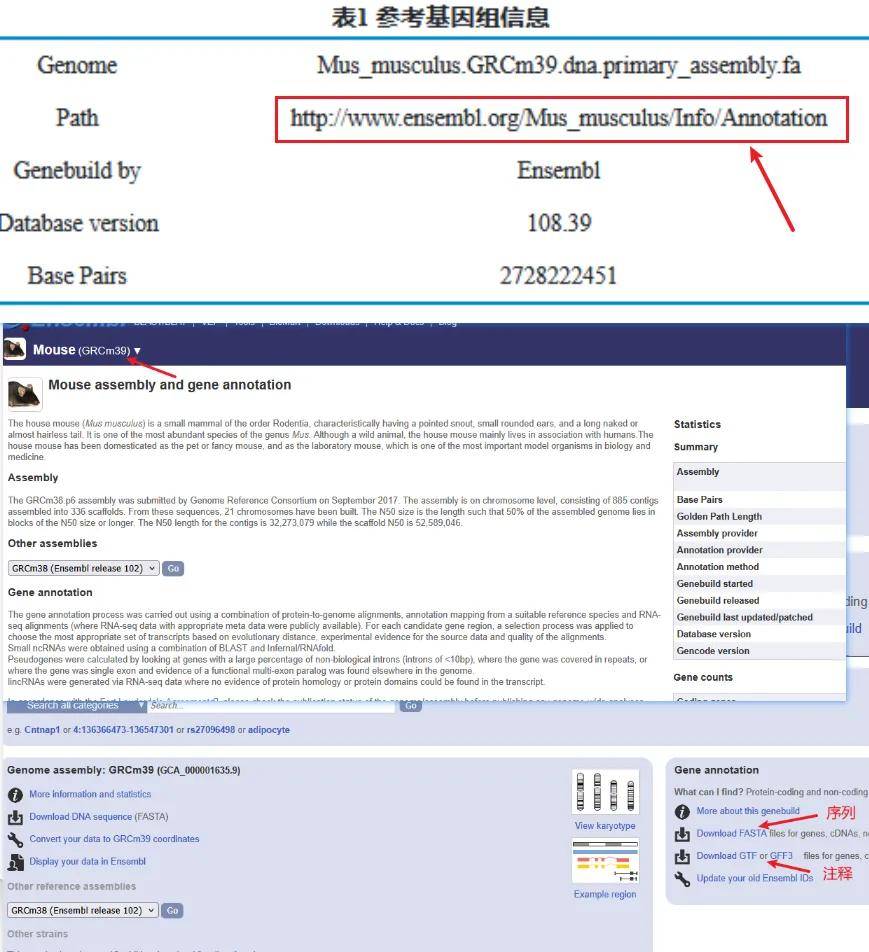
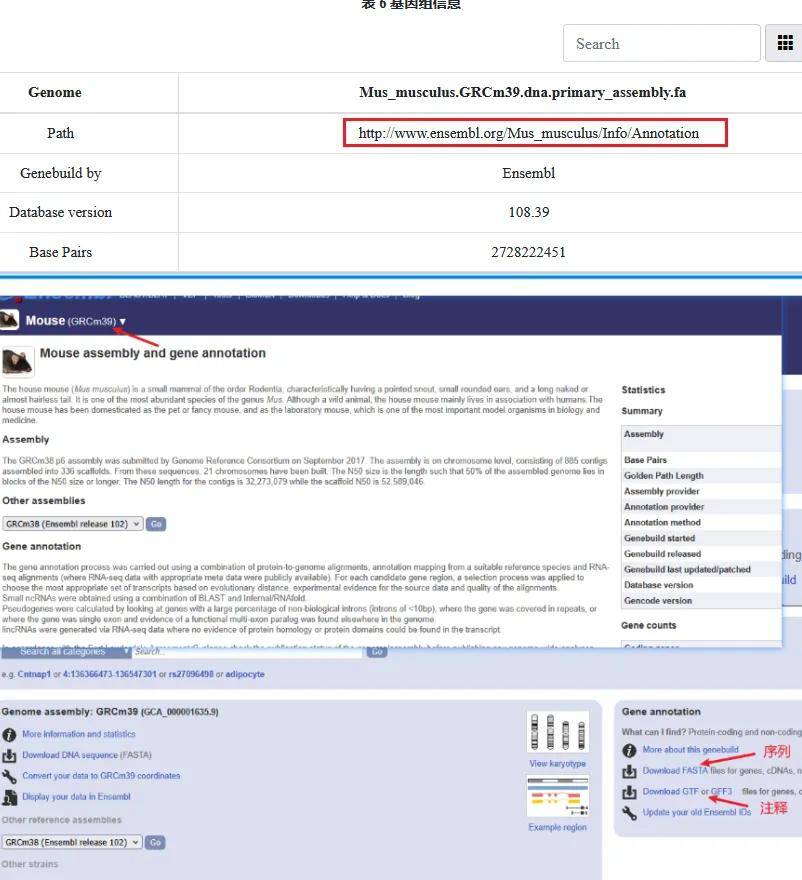
参考基因组的序列文件和结构注释gtf/gff文件:在参考基因组链接(报告参考基因组章节有对应基因组链接)里进入下载。备注:如果是人和小鼠的基因组的话,由于结构注释文件太大,建议下载对应染色体的gff文件查看。
如果分组表填写的参考基因组为链接1(人)、3(小鼠)、5(大鼠)、11(绵羊)、99(水稻)的话,可以在网盘下载对应的序列和注释文件:https://pan.baidu.com/s/1COrNvnK35f3X9ZKPtItOag?pwd=ahrf
如果是原核的项目,参考基因组的序列文件和结构注释gtf/gff文件在原始数据annotation文件夹(云)或report→result→Ref_Genome文件夹(线下)获取。
b.导入IGV
点击 Genomes → Load Genome From File 导入基因组序列文件;点击 File → Load from File 导入基因注释gtf/gff和bam/bw文件。

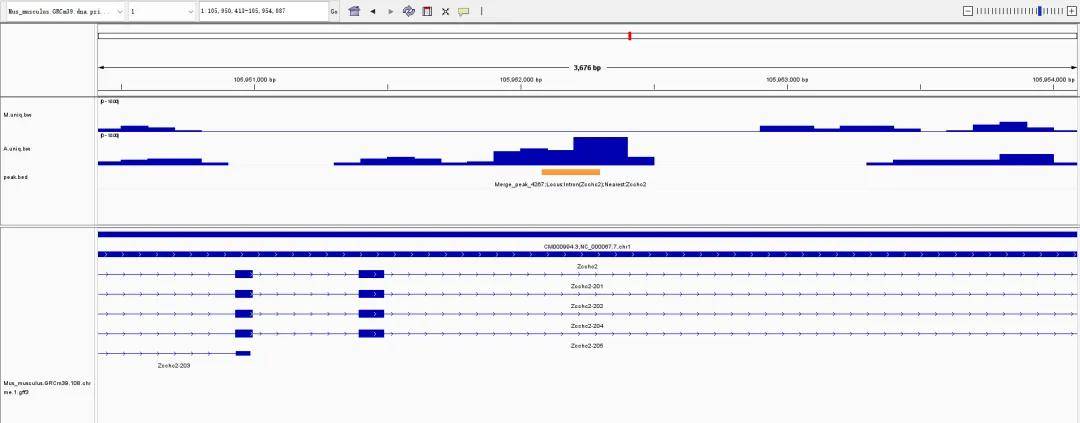
导入成功后如下。可以在bw文件里看到reads覆盖情况,在bam文件中查看reads覆盖、剪切情况、突变情况:
Junctions里可以通过右键 → Sashimi Plot 来实现可变剪切可视化;
放大到一定程度可以查看到序列信息,同时也可以查看发生的插入或突变的具体情况:每个碱基拥有特定的颜色,可以看出从颜色和碱基上看到突变情况;插入的碱基用 Ⅰ 表示,缺失用 - 表示。

3、表观组学peak峰图查看
a.文件准备
参考基因组的序列文件和结构注释gtf/gff文件、样品对应的bw文件(在report→Result→03_MapQC→IGV里)。
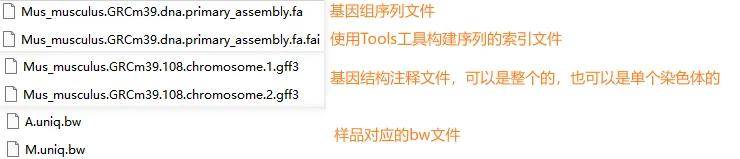
参考基因组的序列文件和结构注释gtf/gff文件:在参考基因组链接(报告参考基因组章节有对应基因组链接)里进入下载。备注:如果是人和小鼠的基因组的话,由于结构注释文件太大,建议下载对应染色体的gff文件查看。
如果分组表填写的参考基因组为链接1(人)、3(小鼠)、5(大鼠)、11(绵羊)、99(水稻)的话,可以在网盘下载对应的序列和注释文件:https://pan.baidu.com/s/1COrNvnK35f3X9ZKPtItOag?pwd=ahrf
b.导入IGV
点击 Genomes → Load Genome From File 导入基因组序列文件;点击 File → Load from File 导入基因注释gtf/gff和bw文件。
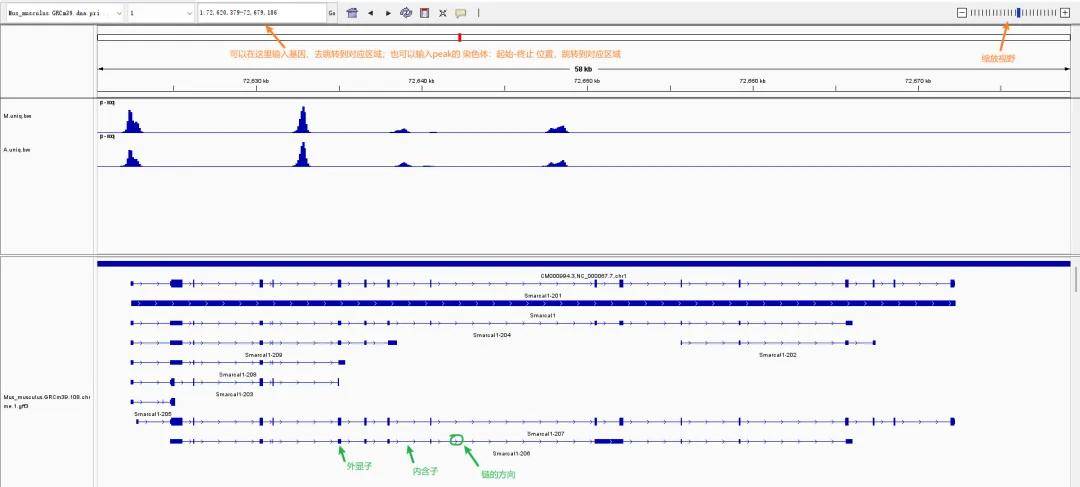
导入成功后如下。可以在bw文件里看到各个区域reads覆盖情况。
c.peak位置导入
因为peak位置在样品或合并结果中并不是如基因一样是固定的,因此我们可以将peak的位置导入到IGV中方便查看。
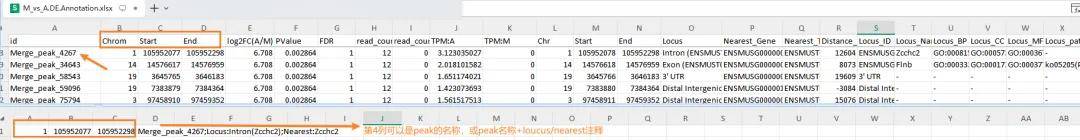
首先在样品peak/合并后的peak/比较组差异peak表里,筛选关注的peak(或所有的peak),整理4项数据到新的表里(染色体,peak起始位置,peak终止位置,标注信息;标注信息可以是peak的名称,也可以整合peak名称和loucus/nearest注释;该表不需要表头)。
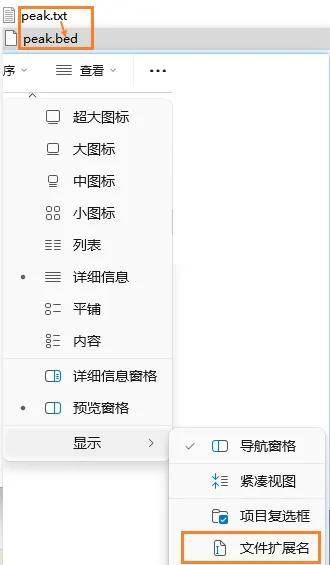
然后将这4列信息复制到txt文件中,在文件夹查看的显示里打开文件扩展名,将txt文件修改后缀为bed文件。
然后在IGV软件中点击 File → Load from File 导入bed文件,通过查找位置,找到关注peak的区域。