

2017-03-15
以PacBio公司的SMRT单分子实时测序技术(Single molecule real-time sequencing)为代表的三代测序技术,通过其独有的环形一致性测序模式(Circular-consensus sequence,CCS),极大提高单碱基测序的准确率,远超Illumina等二代测序技术。与传统转录组测序项目相比,利用PacBio平台的全长转录组测序技术可以直接获得mRNA的全长,保证了mRNA序列的精确性。近期我们将陆续推出全长转录组测序技术相关文章,供讨论和交流。我们在上一期向大家介绍了全长转录组测序的分析内容,本期将为大家对派森诺特色分析内容——可变剪切模型可视化进行重点讲解。
可变剪切模型可视化
可变剪切是造成基因信息多样化的一个重要原因,通过对RNA序列测序可以检测到基因的不同可变剪切模式。今天为大家介绍一种可以将可变剪切模型绘制成图片的Python软件SpliceGrapher。
SpliceGrapher可以根据测序序列预测可变剪切模型,也可以用已知的基因注释文件生成可变剪切模型。它需要两种输入文件,注释gtf/gff3文件和测序reads与参考基因组比对的sam文件,如果有EST序列,也可以用上。
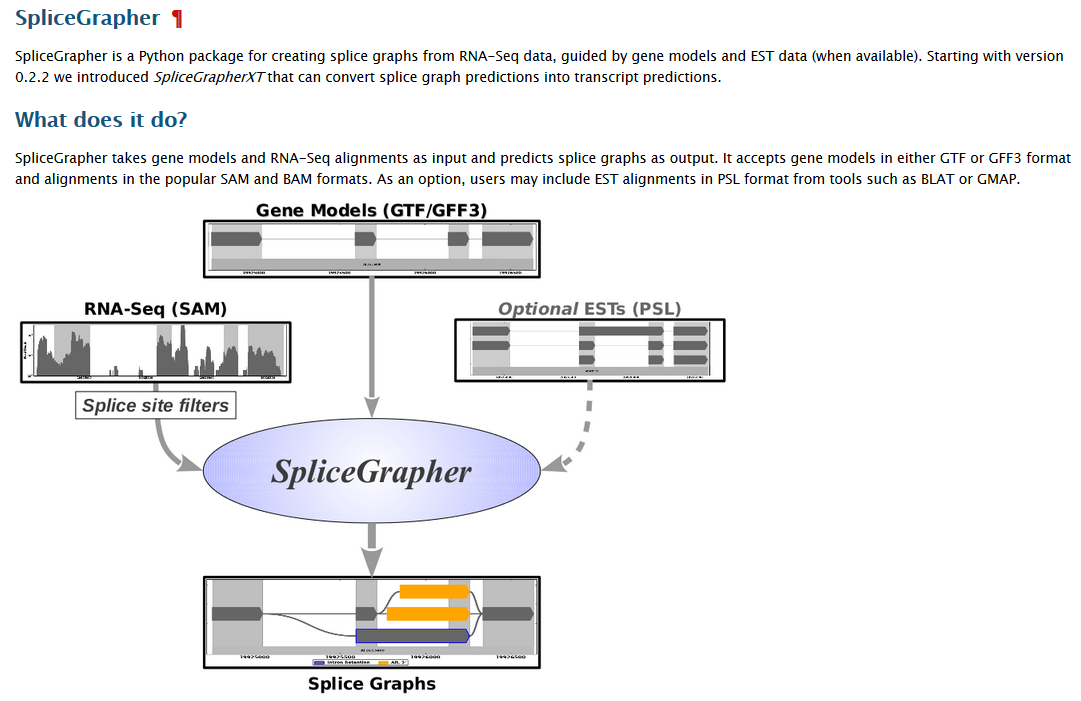
绘制可变剪切模型
SpliceGrapher的使用过程非常简单。首先需要去掉sam文件中的假阳性可变剪切。这里需要用上SpliceGrapher专门准备的物种特性筛选文件,在SpliceGrapher文件夹下有上百种物种的特性筛选文件。然后用筛选后的sam文件预测可变剪切模型,用注释gtf/gff3文件产生已知的可变剪切模型。最后用SpliceGrapher的绘图脚本将可变剪切模型画成图片。
可变剪切图解读
结果图蕴含了许多信息,下面就为大家介绍一下。
一个图片中仅有一个基因的模型,左下角和右下角的数字分别代表该基因的起始位点和终止位点。图片一共分4行,第一行为以该基因的注释文件作出的基因模型,第二行为根据测序结果与注释文件共同作出的基因模型,第三行也是根据测序结果与注释文件共同作出的基因模型,与第二行不同的是只是画出具有代表性的isoform,第四行则是测序文件中支持各外显子的reads数目。
灰色的五边形代表外显子,它们之间的连线表示不同的剪接方式。紫色背景表示有外显子出现的区域(这其中包括保留的内含子),白色背景表示没有外显子出现的区域(即内含子区域)。图中白色背景的宽度并不代表真实的内含子长度,由于有的基因内含子区域远比外显子区域长,为了更清楚地展示可变剪切模型,内含子区域会被缩短很多。
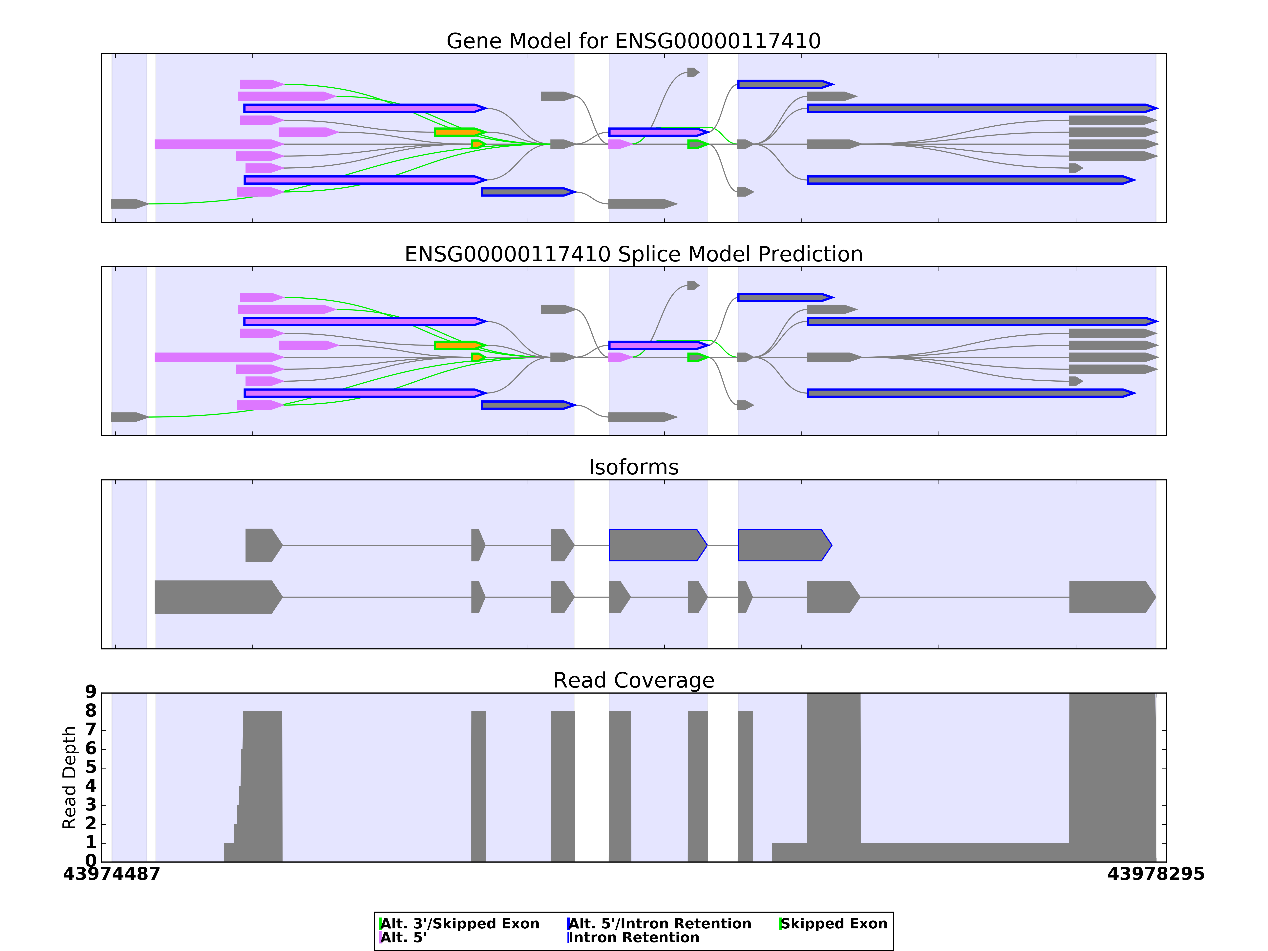
SpliceGrapher可以预测出多种多样的可变剪切事件,已知的基因模型中存在Alt 3’(可变3’端)、Skipped Exon(跳过外显子)、Intron Retention(内含子保留)、Alt 5’(可变5’端)、既是Alt 3’又是Skipped Exon、既是Alt 5’又是Intron Retention等多种可变剪切事件。不同的事件用不同的颜色标注出来,并在图片下方的图例中给予解释。
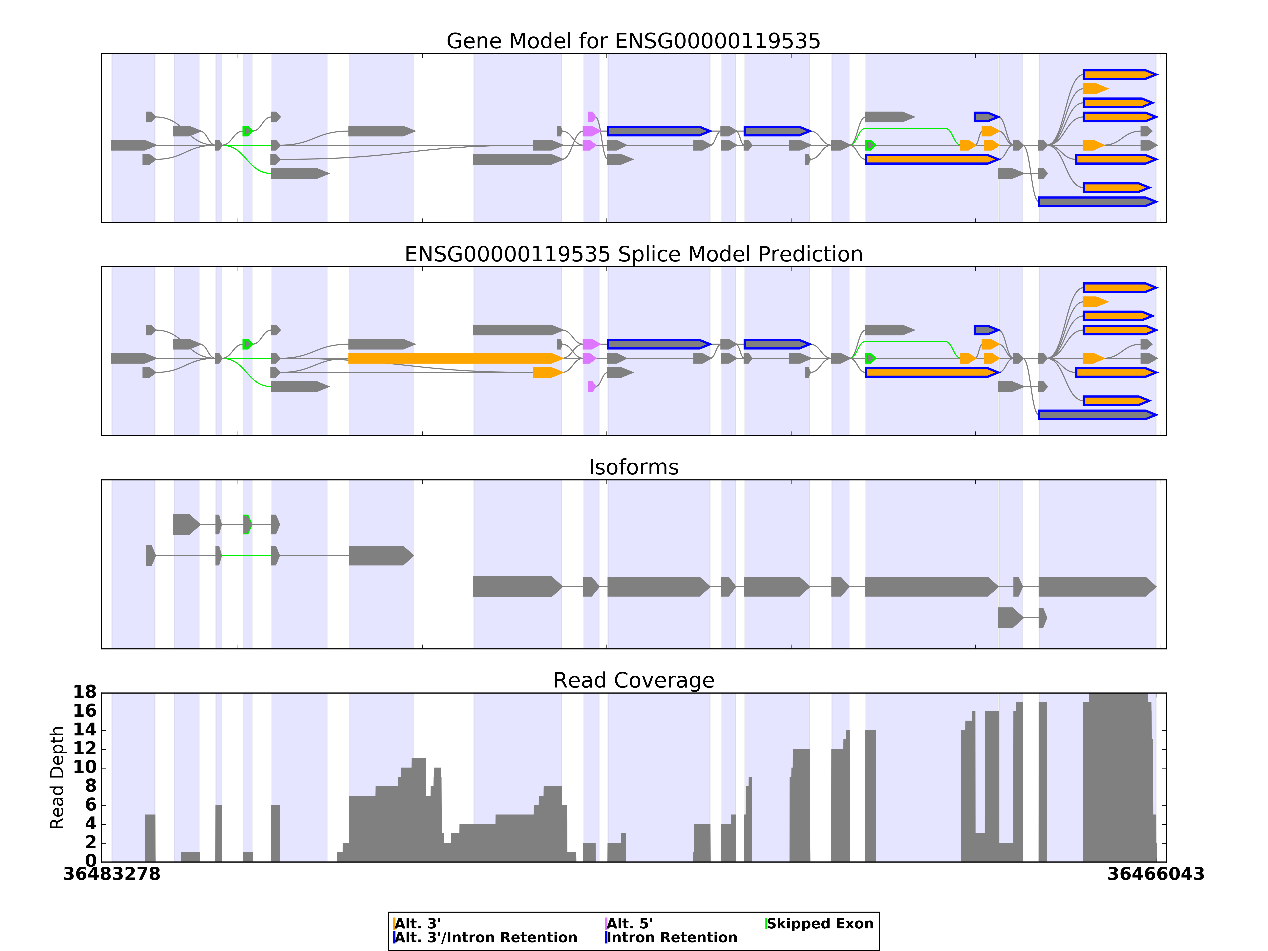
SpliceGrapher预测出的可变剪切模型可能会与已知的基因模型不同,如下图,根据测序结果预测出的基因模型比已知的基因模型多了一个外显子,横跨第6、7个外显子区域和第6个内含子区域。这一点从Read Coverage上也能看出来,在第6个内含子区域有不少reads覆盖度。这也许是一个从未被发现的exon。
好了,关于SpliceGrapher的介绍就到这里了。欢迎大家订购本公司的全长有参转录组分析产品,我们的可变剪切预测项目中会为大家画出物种所有的基因可变剪切模型,并进行分类梳理。


















