

2025-05-28

GO/KEGG富集,每个人都做过,但富集结果中有很多条目,每个条目中又有很多参数需要展示(比如富集到的基因数,P值,richfactor等),如果用常规的Barplot, Dotplot来展示的话,就需要多张图片分别展示部分信息,就会显得图片比较繁杂。那有没有能一张图就展示完所有上述参数信息,又比较美观的图呢?有的兄弟,有的。富集分析圈图就是你要找的图。
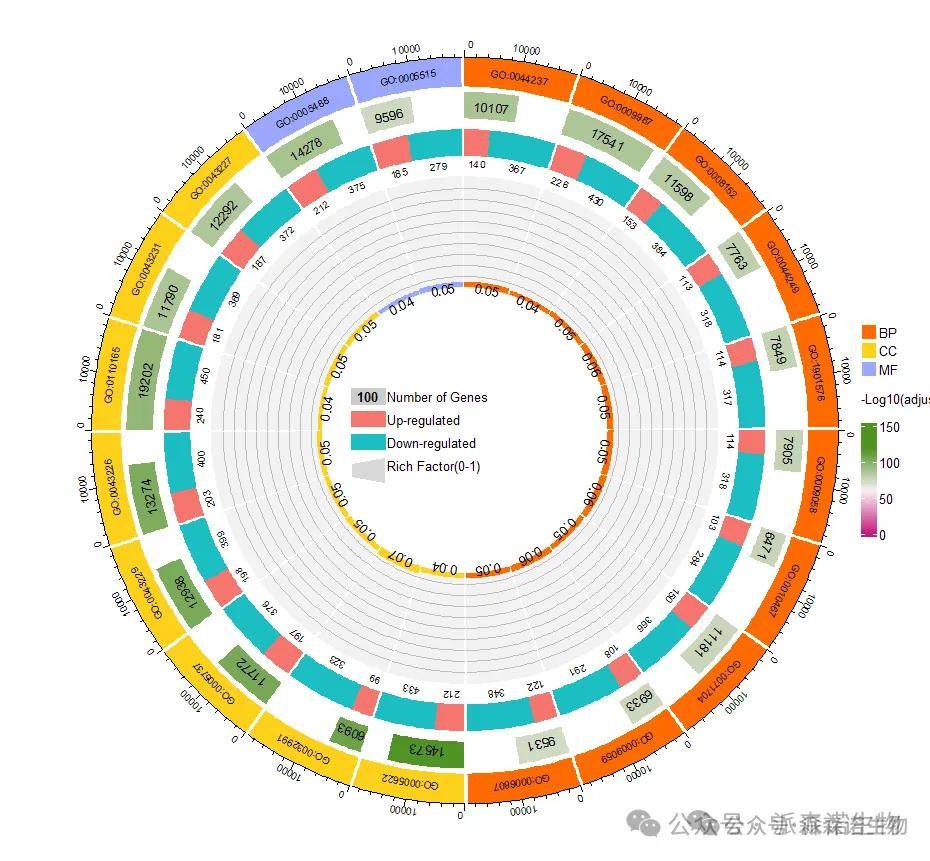
图1 富集分析圈图
图1中展示的就是富集分析圈图,由4层环构成,圆圈中心和右侧是各层的图例。从内圈到外分别展示了:
1.richfactor: 最圈代表富集倍数,在0-1范围内取值,值越大格数越多,颜色代表GO/KEGG类别;
2. 分组基因数:第二圈颜色代表上下调基因,数字代表基因数量;
3. 富集到的基因数与P值: 第三圈代表-log10(pvalue)值,颜色深浅代表值的高低;
4. 通路id及分类:第四圈代表GO/KEGG条目ID,颜色代表GO/KEGG类别。
下面,我们来一层一层地画这张图。
1.数据准备:从富集结果中提取每个Term生成下面这张表格,在R中读取它
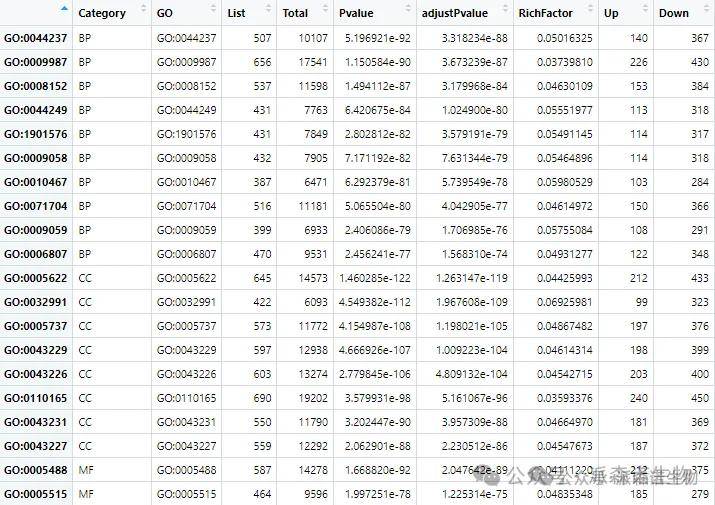
2.第一圈:我们根据表格中的通路分类给通路涂上不同的颜色,并且用通路总基因数给圈图加上坐标。
library(circlize)
library(grid)
library(gridBase)
library(graphics)
library(ComplexHeatmap)
circlize_df <-read.csv(‘富集结果表格’)
circlize_df$Category_color<-color[as.numeric(factor(circlize_df$Category))]
rownames(circlize_df)<-circlize_df$GO
circlize_df$gene_num.min <- 0
circlize_df$gene_num.rich <- circlize_df$Total
circlize_df$gene_num.max <- max(circlize_df$gene_num.rich)
circlize_df$stat_val <- -log10(circlize_df$adjustPvalue)
title_second_data <- "-Log10(adjust.Pvalue)"
circle_size <- unit(1, "snpc")
circos.par(gap.degree = 0.5, start.degree = 90)
plot_data <- circlize_df[, c("GO", "gene_num.min", "gene_num.max")]
plot.new()
pushViewport(viewport(
x = 0, y = 0.5, width = circle_size, height = circle_size,
just = c("left", "center")
))
par(omi = gridOMI(), new = TRUE)
circos.genomicInitialize(plot_data, plotType = NULL)
circos.track(
ylim = c(0, 1), track.margin = c(0, 0), track.height = 0.08, bg.border = NA, bg.col = circlize_df$Category_color,
panel.fun = function(x, y) {
ylim <- get.cell.meta.data("ycenter")
xlim <- get.cell.meta.data("xcenter")
sector.name <- get.cell.meta.data("sector.index")
circos.axis(h = "top", labels.cex = 0.6, labels.pos.adjust = T, labels.col = "#000000", labels.niceFacing = FALSE)
circos.text(xlim, ylim, sector.name, cex = 0.6, col = "#000000", niceFacing = FALSE)
}
)
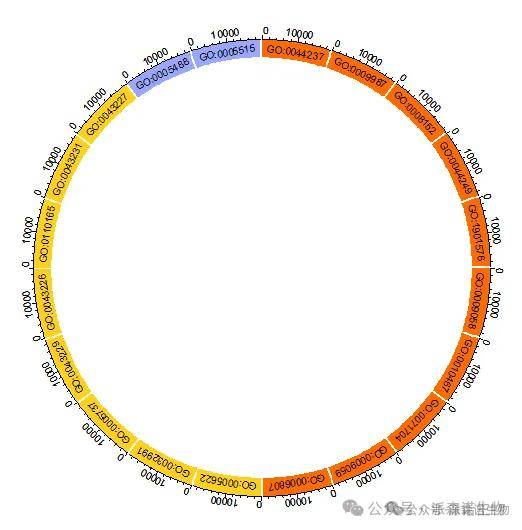
图2 富集分析圈图的第一圈
3.第二圈:绘制富集的基因和富集p值。
plot_data <- circlize_df[, c("GO", "gene_num.min", "gene_num.rich", "stat_val")]
label_data <- circlize_df[, "gene_num.rich"]
label_data <- as.data.frame(label_data)
rownames(label_data) <- circlize_df$GO
p_max <- round(max(circlize_df[["stat_val"]])) + 1
colorsChoice <- colorRampPalette(second_col)
color_assign <- colorRamp2(breaks = 0:p_max, col = colorsChoice(p_max + 1))
circos.genomicTrackPlotRegion(
plot_data,
track.margin = c(0, 0), track.height = 0.1, bg.border = NA, stack = TRUE,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...)
ylim <- get.cell.meta.data("ycenter")
xlim <- label_data[get.cell.meta.data("sector.index"), 1] / 2
sector.name <- label_data[get.cell.meta.data("sector.index"), 1]
circos.text(xlim, ylim, sector.name, cex = 0.8, col = "#000000", niceFacing = FALSE)
}
)
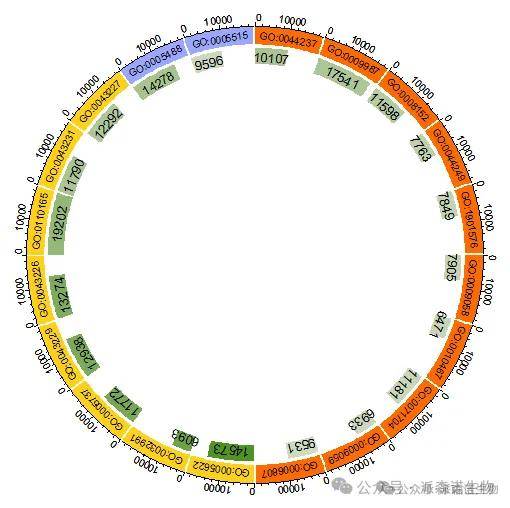
图3 添加富集分析圈图的第二圈
4.第三圈:分组基因数。这里我们用上下调基因作为分组。
circlize_df$up.proportion <- circlize_df$Up / (circlize_df$Up+circlize_df$Down)
circlize_df$down.proportion <- circlize_df$Down / (circlize_df$Up+circlize_df$Down)
circlize_df$up_v <- circlize_df$up.proportion * circlize_df$gene_num.max
plot_data_up <- circlize_df[, c("GO", "gene_num.min", "up_v")]
names(plot_data_up) <- c("id", "start", "end")
plot_data_up$type <- 1
circlize_df$down_v <- circlize_df$down.proportion * circlize_df$gene_num.max + circlize_df$up_v
plot_data_down <- circlize_df[, c("GO", "up_v", "down_v")]
names(plot_data_down) <- c("id", "start", "end")
plot_data_down$type <- 2
plot_data <- rbind(plot_data_up, plot_data_down)
label_data <- circlize_df[, c("up_v", "down_v", "Up", "Down")]
label_data <- as.data.frame(label_data)
rownames(label_data) <- circlize_df$GO
color_assign <- colorRamp2(breaks = c(1, 2), col = c(upcol, downcol))
circos.genomicTrackPlotRegion(
plot_data,
track.margin = c(0, 0), track.height = 0.1, bg.border = NA, stack = TRUE,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...)
ylim <- get.cell.meta.data("cell.bottom.radius") - 0.5
xlim <- label_data[get.cell.meta.data("sector.index"), 1] / 2
sector.name <- ifelse(label_data[get.cell.meta.data("sector.index"), 3] > 0, label_data[get.cell.meta.data("sector.index"), 3], " ")
circos.text(xlim, ylim, sector.name, cex = 0.6, col = "#000000", niceFacing = FALSE)
xlim <- (label_data[get.cell.meta.data("sector.index"), 2] + label_data[get.cell.meta.data("sector.index"), 1]) / 2
sector.name <- ifelse(label_data[get.cell.meta.data("sector.index"), 4] > 0, label_data[get.cell.meta.data("sector.index"), 4], " ")
circos.text(xlim, ylim, sector.name, cex = 0.6, col = "#000000", niceFacing = FALSE)
}
)
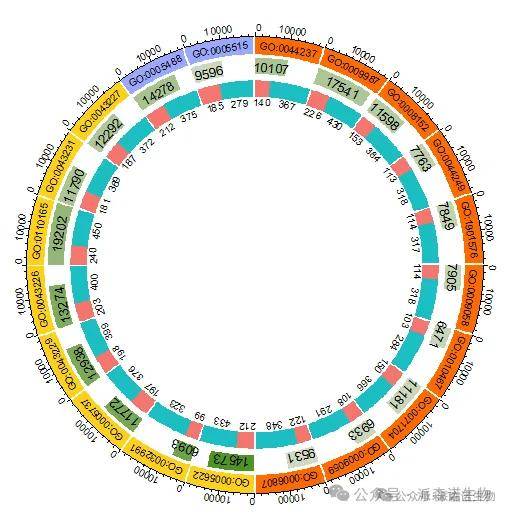
图4 添加富集分析圈图的第三圈
5.第四圈:绘制富集因子。
plot_data <- circlize_df[, c("GO", "gene_num.min", "gene_num.max", "RichFactor")]
label_data <- circlize_df[, c("Category", "RichFactor")]
label_data$RichFactor <- round(label_data$RichFactor, 2)
label_data <- as.data.frame(label_data)
rownames(label_data) <- circlize_df$GO
circos.genomicTrack(
plot_data,
track.margin = c(0.01, 0.04), track.height = 0.3, ylim = c(0.05, 0.95), bg.col = "gray95", bg.border = NA, # ylim = c(0, 1)
panel.fun = function(region, value, ...) {
sector.name <- get.cell.meta.data("sector.index")
for (i in seq(0.1, 0.9, by = 0.1)) {
circos.lines(c(0, max(region)), c(i, i), col = "gray", lwd = 0.3)
}
circos.genomicRect(region, value, col = circlize_df[sector.name,'Category_color'], border = NA, ytop.column = 1, ybottom = 0, ...)
ylim <- label_data[get.cell.meta.data("sector.index"), 2] - 0.05
xlim <- get.cell.meta.data("xcenter")
sector.name <- label_data[sector.name, 2]
circos.text(xlim, ylim, sector.name, cex = 0.8, col = "#000000", niceFacing = FALSE)
}
)
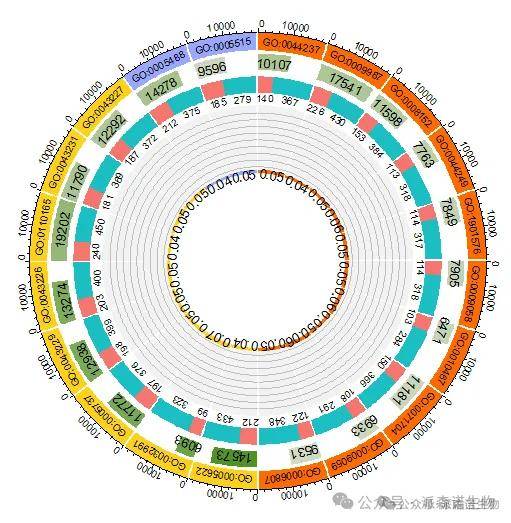
图5 添加富集分析圈图的第四圈
6.最后一步:绘制图例。
updown_legend <- Legend(
labels = c("Number of Genes", "Up-regulated", "Down-regulated", "Rich Factor(0-1)"),
graphics = list(
function(x = 0, y, w, h) {
grid.draw(gTree(
children = gList(
rectGrob(x = 0, y, w * 2.2, h * 1, gp = gpar(fill = "gray80", col = "gray80")),
textGrob("100", x = 0, y)),
gp = gpar(col = "black", fontsize = 10, fontface = "bold")))},
function(x = 0, y, w, h) {
grid.rect(x = 0, y, w * 2.2, h * 1, gp = gpar(fill = upcol, col = upcol))},
function(x = 0, y, w, h) {
grid.rect(x = 0, y, w * 2.2, h * 1, gp = gpar(fill = downcol, col = downcol))},
function(x = 0, y, w, h) {
grid.polygon(y = c(0.09, -0.05, -0.12, 0.16), x = c(-0.13, -0.13, 0.13, 0.13), gp = gpar(fill = "gray85", col = "gray85"))}),
labels_gp = gpar(fontsize=10), # grid_height = unit(0.6, 'cm'), grid_width=unit(0.8, 'cm'),
row_gap = unit(2, "mm"))
category_legend <- Legend(
labels = unique(circlize_df$Category),
type = "points", pch = NA, background = unique(circlize_df$Category_color),
labels_gp = gpar(fontsize = 10), row_gap = unit(1, "mm")
) # grid_height = unit(0.6, 'cm'), grid_width = unit(0.8, 'cm'))
pvalue_legend <- Legend(
col_fun = colorRamp2(breaks = 0:p_max, col = colorsChoice(p_max + 1)),
legend_height = unit(3, "cm"), labels_gp = gpar(fontsize = 10),
title_gp = gpar(fontsize = 10), title_position = "topleft", title = paste0(title_second_data, "\n"), row_gap = unit(3, "mm"))
lgd_list_vertical <- packLegend(updown_legend)
lgd_list_vertical2 <- packLegend(category_legend, pvalue_legend)
draw(lgd_list_vertical, just = "center")
upViewport()
draw(lgd_list_vertical2, x = circle_size, just = "left")
应用场景与注意事项
1.适用场景:
可用于解析生物学机制,如揭示疾病、发育等过程的关键通路;注释细胞亚群功能,明确其特性;筛选生物标志物;辅助药物靶点发现与重定位;整合多组学数据及指导实验验证方向等,助力生物医学研究。
2.可视化调整:
可根据研究重点调整颜色方案(如按通路类别分组着色)、过滤低富集因子的条目,或添加热图刻度条增强可读性。
总 结
富集圈图通过多维度信息的空间整合,为复杂的基因功能分析提供了直观的解读框架,助力研究者快速定位关键通路与核心基因,推动单细胞转录组数据的深度挖掘。绘制图片或者复现代码过程中,如果老师遇到疑惑,欢迎拨打我们的热线电话或者联系我们的驻地销售。


















