




















Highlights
1、传统医疗的“千人一方”模式在复杂疾病(如肿瘤、代谢综合征)中面临显著局限,机器学习赋能多组学可实现精准医疗破局。
2、多组学+机器学习的强强联合已被广泛应用到精准医疗领域,如癌症早期筛查、癌症分子分型、免疫治疗靶点发现等。
3、派森诺微生物组、代谢组等组学及相关联合分析可以进行随机森林、支持向量机、XGBoost、朴素贝叶斯、逻辑回归、套索回归和多模型联合等多种机器学习分析。
传统医疗依赖症状和单一指标等进行诊疗,对疾病分子机制缺乏深入理解。这种“千人一方”模式在复杂疾病(如肿瘤、代谢综合征)中面临显著局限,常导致诊断滞后与疗效欠佳。而精准医疗通过整合微生物组、转录组、代谢组等多维度生物分子网络解析,结合个体遗传背景与环境暴露特征,构建"分子分型-动态监测-靶向干预"的新型诊疗体系,实现从粗放式医疗向分层化、预防性、个体化医疗模式的转变。从而达到精准预防、精准诊断以及精准治疗。

但多组学从数据分析到临床决策过程中面临数据异质性、因果验证、临床转化三大挑战。多组学数据的高维度、异质性和技术噪声是精准医疗落地的核心障碍。机器学习可通过多模态数据融合,显著提升数据质量与生物学意义的提取效率;还可通过深度特征学习和因果关系建模,突破传统统计方法的局限,揭示疾病分子机制等。机器学习不仅提升了多组学分析的效率与准确性,更为多组学在精准医疗中提供新的赋能,实现更早的诊断窗口期、更精准的治疗策略、更低的医疗成本。在可见的将来,随着多组学、时空组学与机器学习算法的深度融合,精准医疗将实现从 “疾病治疗” 到 “健康管理” 的终极目标,推动个体化医疗普惠化进程。
在多组学中,微生物组与代谢组的联合分析是解锁微生物与宿主之间复杂相互作用的关键手段之一,它为精准医疗等医学研究与应用开辟了新的道路。接下来跟着小派来看看机器学习如何赋能微生物组和代谢组,从而实现精准医疗的破局!
案例一
Integrated metagenomic and metabolomic analysis reveals distinctive stage-specific gut-microbiome-derived metabolites in intracranial aneurysms
综合宏基因组学和代谢组学分析揭示了颅内动脉瘤中独特的阶段特异性肠道微生物衍生代谢产物
期刊:Gut(IF:23.1 )
研究背景
颅内动脉瘤破裂缺乏有效的风险评估工具,因此探索肠道微生物群及其代谢产物对颅内动脉瘤进展和破裂的影响具有重要意义。本研究聚焦于颅内动脉瘤(IA)的形成与破裂机制,特别是肠道微生物及其代谢产物在这一过程中可能扮演的角色。
样本
宏基因组:59名UIA患者和50名RIA患者的粪便样本;
代谢组学:30名RIA患者和30名UIA患者的血浆;
靶向代谢组学:
162名UIA患者和97名RIA患者作为发现队列;
49名RIA患者和195名UIA患者作为独立验证队列。
流程图
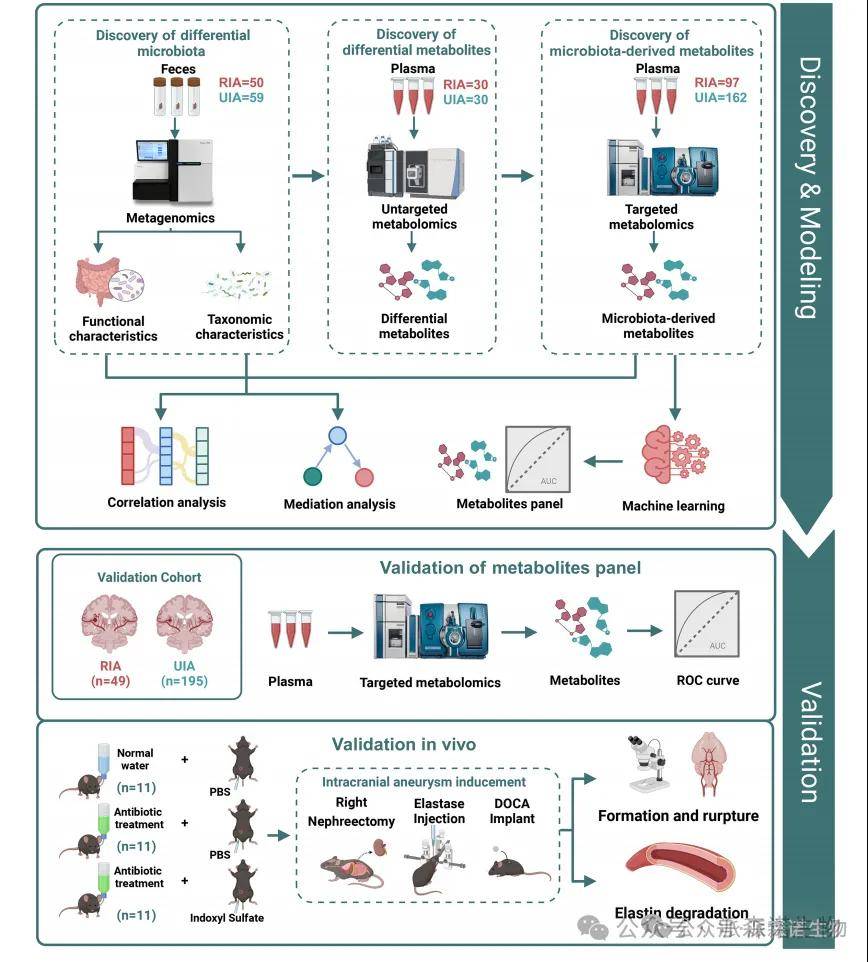
结 果
1、在 RIA 患者的粪便样本中,肺炎克雷伯菌和瘤胃球菌属的含量明显增加。氨基酸和吲哚化合物等33种代谢物在RIA患者血浆中显著增加;
2、靶向代谢组学分析,RIA和UIA组之间胆汁酸和色氨酸代谢物存在显著差异。随机森林模型构建了一个由硫酸吲哚酚(IS)、5-羟色氨酸和犬尿氨酸组成的代谢物组合,该组合可以区分RIA患者和UIA患者,其在测试集中的组合AUC为0.974,在外部验证集中为0.972,这一代谢物组合或可用于发现IA进展的发病机制,甚至预测动脉瘤的破裂。
3、基于相关性分析表明,IS水平的升高与色氨酸代谢途径和肺炎克雷伯菌密切相关。
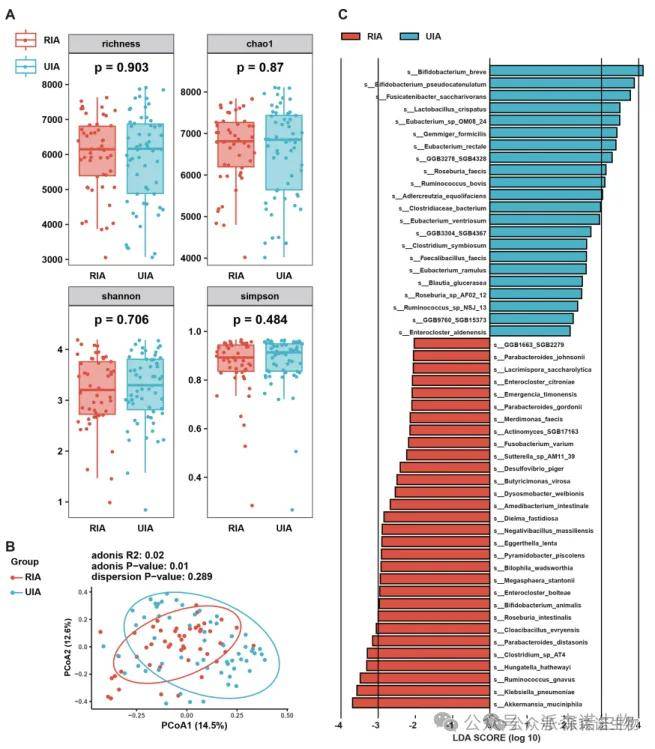 粪便宏基因组学
粪便宏基因组学
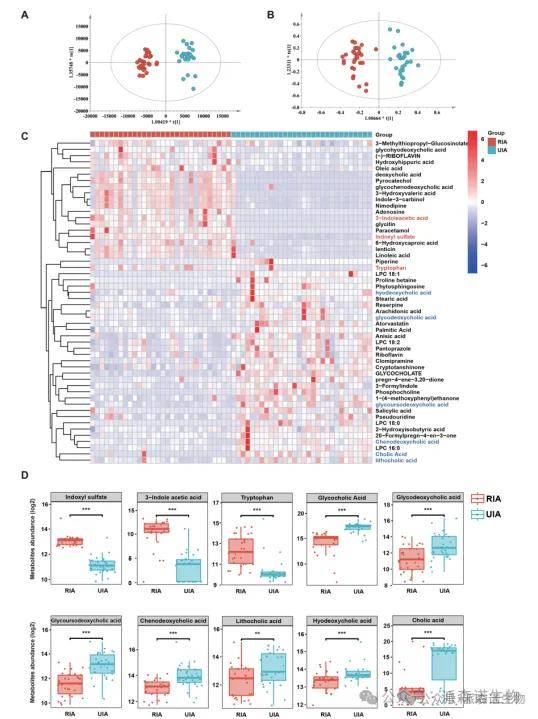 非靶向代谢组学
非靶向代谢组学
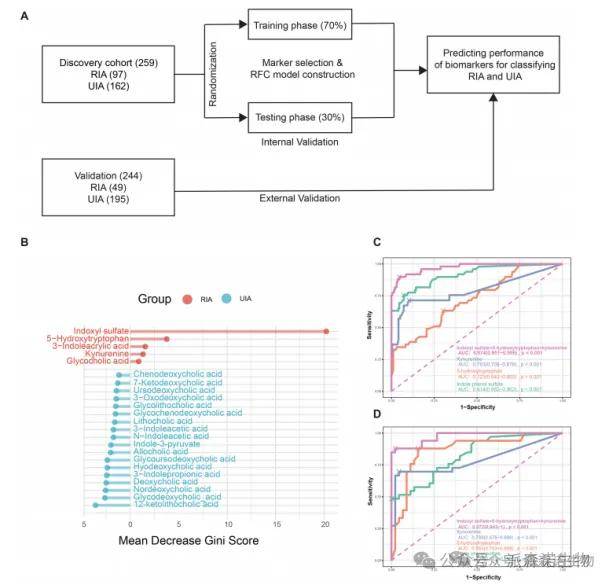 随机森林模型筛选标志物
随机森林模型筛选标志物
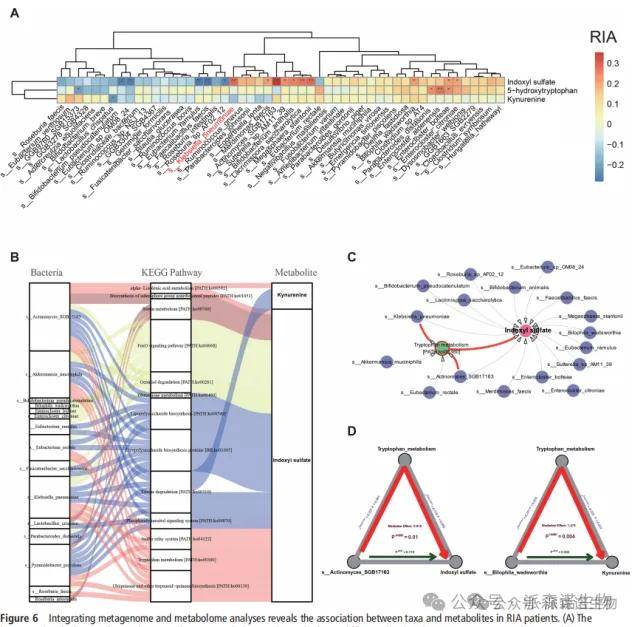 宏基因组和代谢组关联
宏基因组和代谢组关联
案例二
Integrated metagenomic and metabolomic analysis reveals distinct gut-microbiome-derived phenotypes in early-onset colorectal cancer
综合宏基因组和代谢组学分析揭示了早发性结直肠癌癌症中不同的肠道微生物衍生表型
期刊:Gut(IF:23.1 )
研究背景
早发性结直肠癌(EO-CRC)全球发病率持续攀升。由于常规筛查通常从50岁开始,年轻患者极易漏诊,这导致 EO-CRC 患者的病理特征更具侵袭性,且缺乏针对性的治疗方案。目前,EO-CRC 患者是否存在独特的分子特征和多组学图谱尚不明确。与此同时,大量研究表明肠道微生物群及其代谢物在结直肠癌发病机制中起着关键作用。基于此,本文提出假设:年轻个体中肠道微生物群和代谢物的改变可能引发 EO-CRC 的早期发作。
样本
微生物和代谢物检测:
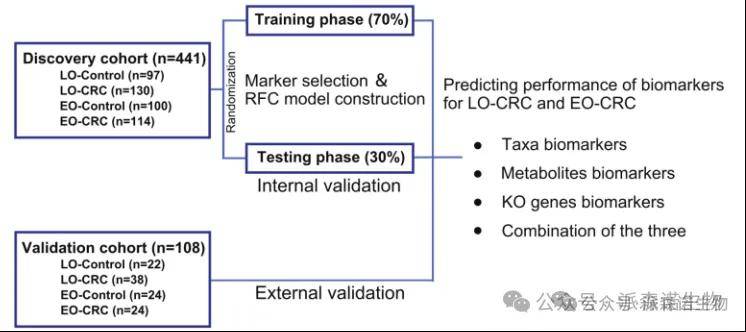
发现集(441例):114名EO-CRC粪便;130名晚发性结直肠癌(LO-CRC)粪便;197名健康受试者粪便。
验证集(108例):24名EO-CRC粪便;38名LO-CRC粪便;46名健康受试者粪便。
流程图
结 果
1、与对照相比,LO-CRC 和 EO-CRC 组的 α 多样性显著降低。PCoA 显示,不同组的微生物群落结构存在显著差异。在CRC组种丰度下降的细菌包括产丁酸细菌普拉梭菌等。
2、氨基酸代谢物,如l -苯丙氨酸和D-鸟氨酸在两种CRC中都显著增加,而一些特定的氨基酸仅在EO-CRC样本中富集。
3、宏基因组KEGG富集结果指示出了氨基酸代谢、不同环境下的微生物代谢和脂质代谢的差异。
4、微生物KO基因和代谢物的改变与CRC粪便中微生物群的变化有关,数据综合分析可能部分解释LO-CRC和EO-CRC的不同发病机制。
5、基于细菌物种组成、代谢物水平和酶基因丰度,使用随机森林方法构建分类器。在测试阶段,单独使用细菌分类群、代谢物或KO基因标记物作为LO-CRC和LO-Control之间的预测因子,产生的AUC分别为84.53%、83.3%和84.97%,然而三者结合后,AUC达到92.34%,该结果在验证集中得到了确认:细菌分类群、代谢物、KO基因的AUC分别为78.17%、78.47%、81.22%。而在EO-CRC中,49个丰度不同的物种、36个代谢物、59个KO基因和27个综合标志物被选为最佳标志物集,且三特征综合模型比单独分析有更准确的区分效果,外部验证队列中也显示出类似的结果。
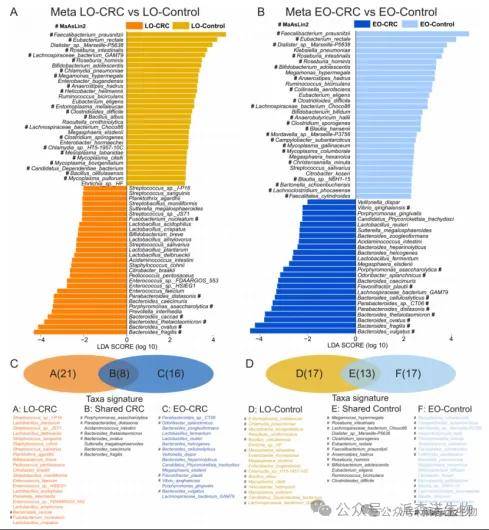 微生物群的分类特征
微生物群的分类特征
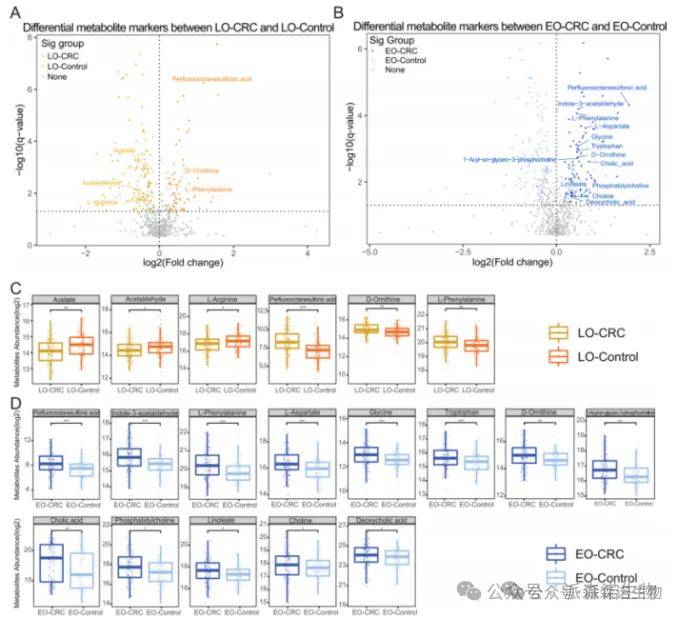 粪便代谢组改变
粪便代谢组改变
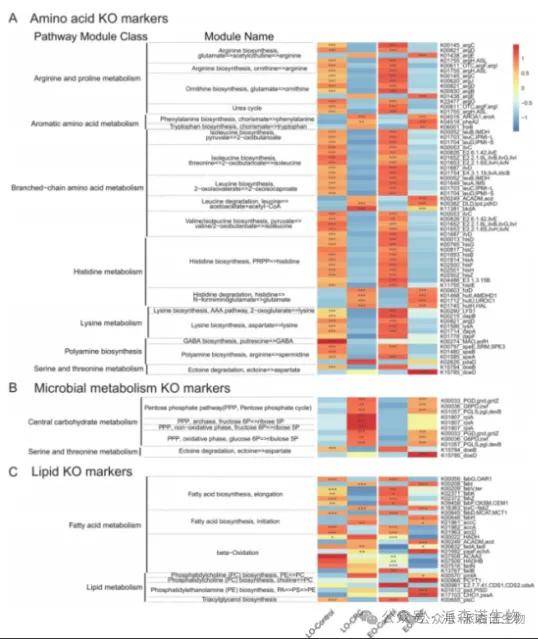 功能富集分析
功能富集分析
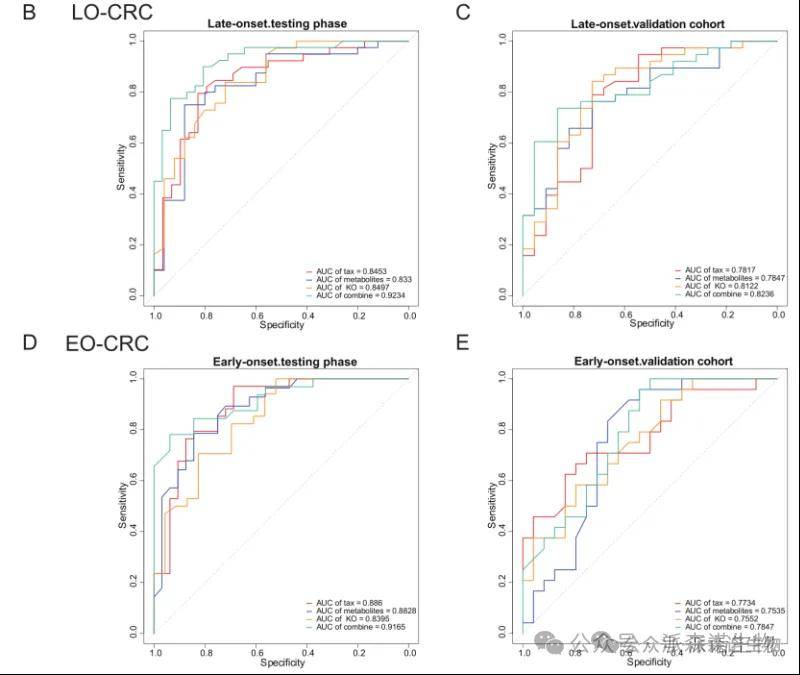 随机森林模型整合多组学标记
随机森林模型整合多组学标记
派森诺机器学习标准分析实例
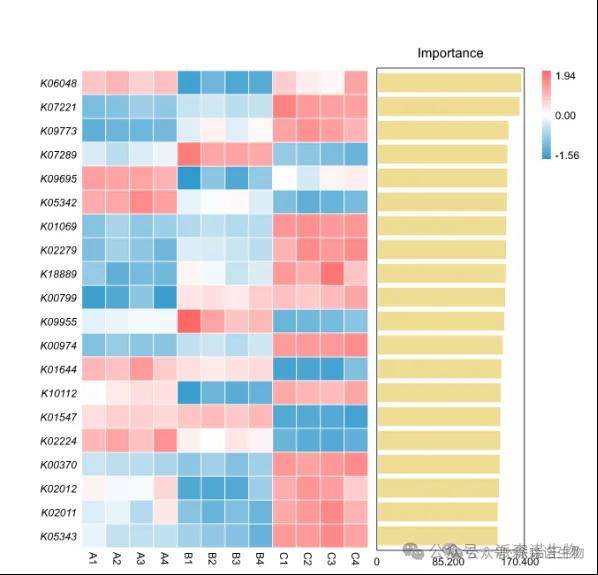 随机森林重要特征图
随机森林重要特征图
 ROC分析
ROC分析
