

2025-06-09

Highlights
1.从传统的GWAS到XGWAS/XWAS,整合多组学数据,突破单一基因组分析的局限,全方位挖掘性状相关机制。
2.一网打尽XGWAS/XWAS开展策略,根据不同关注层面合理选择测序深度和相关组学,确保数据关联准确。
3.派森诺提供多种模型和系统分析流程,涵盖群体遗传结构、全基因组关联分析及高级选择性清除分析,深度解析复杂性状的遗传基础。
全基因组关联分析(GWAS)是一种用于识别遗传区域和性状之间关联的方法,它通过检测多个个体在全基因组范围内的遗传变异多态性,并与可观测的性状(表型)进行群体水平的统计学分析,以筛选出可能影响该性状的遗传变异。如今,科研领域的内卷那是越来越严重,可以发现越来越多的高分文章里面开始频繁出现其他类型的关联分析,而不是仅仅局限于GWAS。我们可以通过整合转录组/蛋白质组关联研究(Transcriptome/Proteome association studies, 简称T/PWAS,统称为XWAS)能够将表型与特定基因的影响联系起来。当然,另一种较为简单的理解或分析方法可以直接将基因/蛋白表达、某类代谢物甚至微生物作为表型,而可以进一步称为XGWAS。
一、常用XGWAS/XWAS关联分析简介
● Genome-Wide Association Study (GWAS)
GWAS通过对众多遗传标记(通常是单核苷酸多态性,SNP)进行挖掘,寻找与特定性状或表型相关的遗传变异。当某些遗传变异在具有特定表型的群体中的频率显著高于另一人群时,这些变异就被认为与该表型可能存在关联。其目的是识别出影响复杂性状的基因位点,帮助揭示其遗传基础。目前,更多变异类型加入其中,如Indel/SV-GWAS更多关注小片段变异/结构性变异与表型的关联。
● Transcriptome-Wide Association Study (TWAS)
全转录组关联研究(TWAS)将基因组信息整合成与功能相关的单位,对应于基因及其表达。GWAS已经成功鉴定了数千个与多种复杂性状相关的遗传变异。然而,对于许多性状往往由多个微效基因控制。而TWAS是研究这些潜在变异-性状关联机制的宝贵工具。TWAS能够将GWAS与表达图谱研究结合起来,从而识别基因-性状关联(GTAs)。
● Proteome-Wide Association Study (PWAS)
全蛋白质组关联研究(Proteome-Wide Association Study,PWAS),用于检测由蛋白质功能改变介导的基因-表型关联。PWAS汇总了共同影响蛋白编码基因的所有变异信号,并利用机器学习及概率模型评估它们对蛋白质功能的整体影响。随后,它检测该基因是否在个体间展现出与目的表型相关的功能性变异。PWAS能够捕捉包括隐性遗传在内的复杂遗传模式。
● metabolome Genome-Wide Association Study (mGWAS)
mGWAS是将代谢组学数据作为表型,与基因组数据进行关联分析的一种领先方法。由于代谢组数据对表型鉴定更为精细,因此关联分析的精度比传统GWAS分析更高,通过代谢组与基因组整合的mGWAS分析,可以得到更精准的代谢物的调控基因信息。
● Microbial Genome-Wide Association Study (MGWAS)
MGWAS是一种用于研究微生物与宿主遗传变异之间关联的方法,尤其在探究植物-微生物互作领域已逐渐成为一种新兴的分析手段,用于探索宿主遗传变异如何影响微生物群落的定植和功能。
二、如何开展 XGWAS/XWAS?
● 材料选择

● 测序策略
1)全基因组重测序。针对有参考基因组,基于关注目标变异类型不同选择合适的测序深入,如SNP和Indel建议测序深度10X以上,SV则建议测序深度20X以上。
2)多组学测定。根据关注层面不同进而选择转录组/蛋白组/代谢组/微生物组,注意:测定个体需与重测序样本一一对应。
● 模型选择
线性模型(GLM)和混合线性模型(MLM)是进行GWAS时常用且经典的两种模型。PS:派森诺除了标准交付的结果,针对关键性状(n<10),可采用4种模型进行定位(压缩混合线性模型(CMLM)和固定和随机模型交替概率统一模型(FarmCPU)等)!
● 分析流程
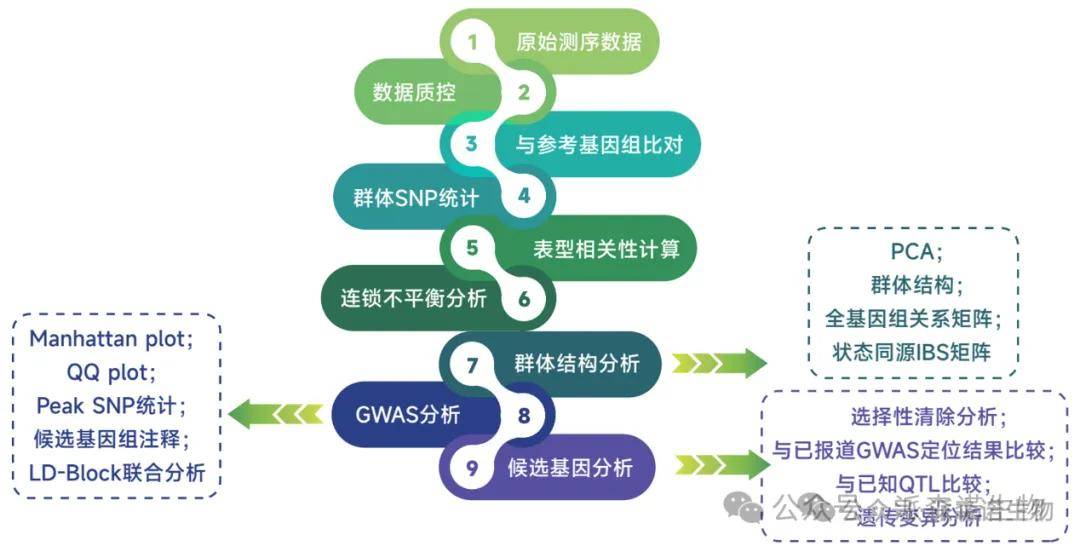
GWAS分析流程
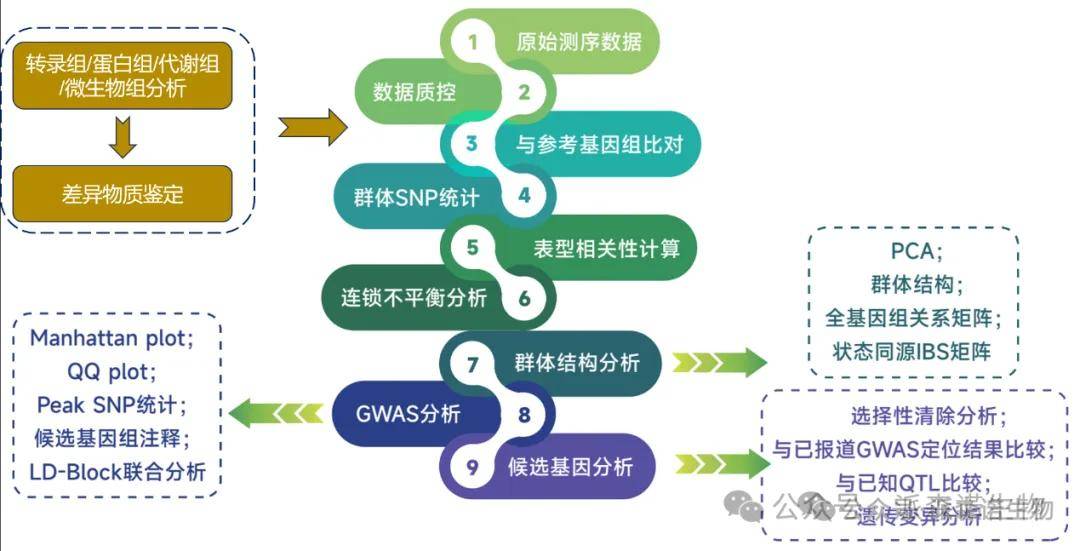
XGWAS分析流程
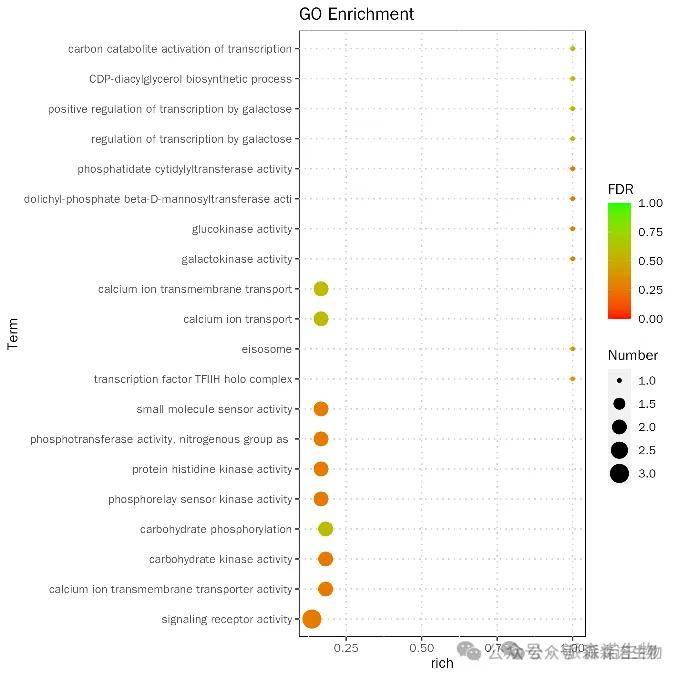
三、XGWAS/XWAS部分核心内容展示
● 群体遗传结构相关分析
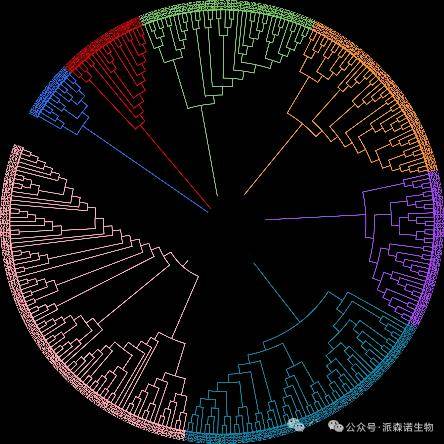
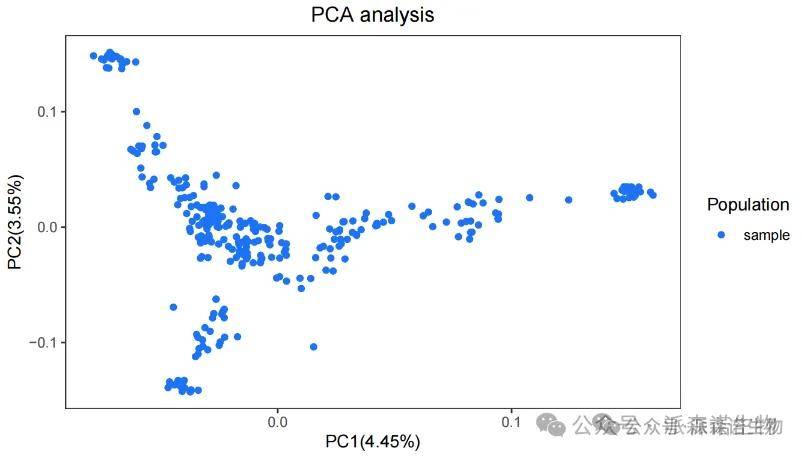
系统进化树重构及群体PCA分析
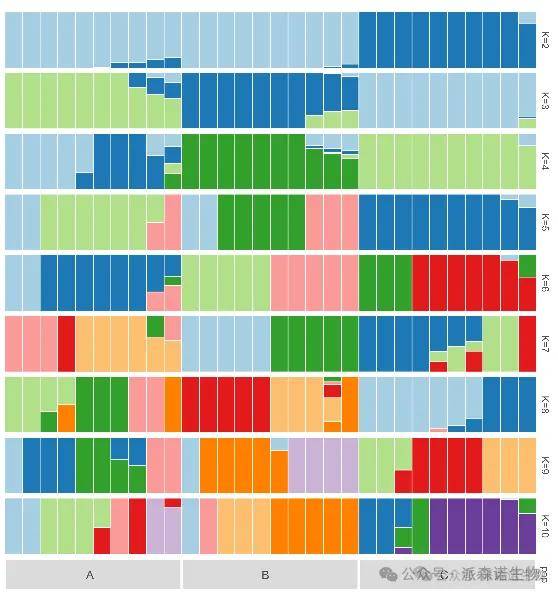
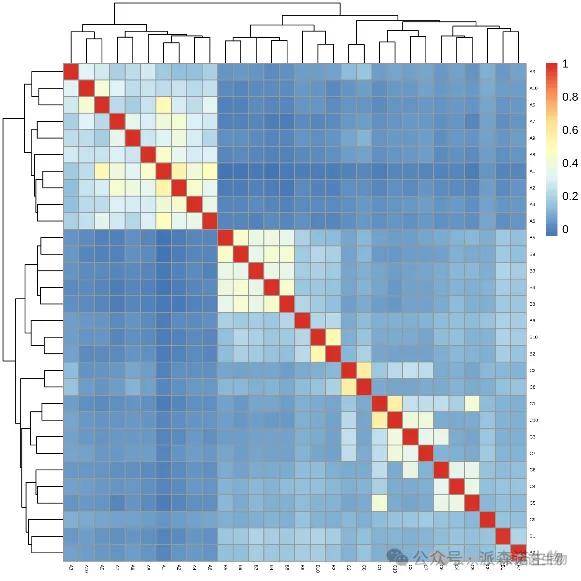
群体遗传结构图及亲缘关系分析
● 全基因组关联分析分析
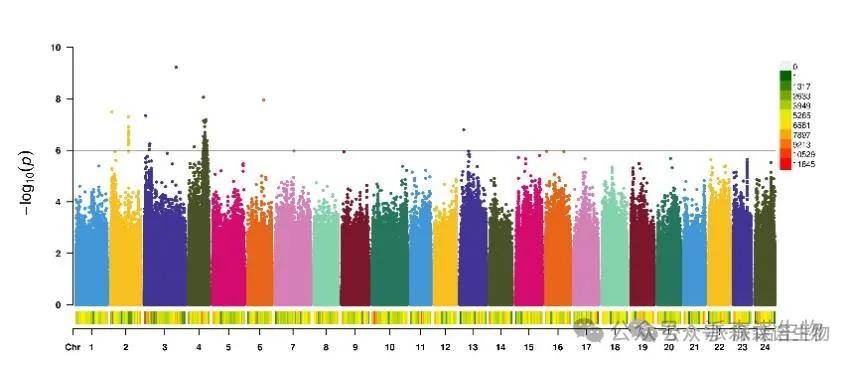
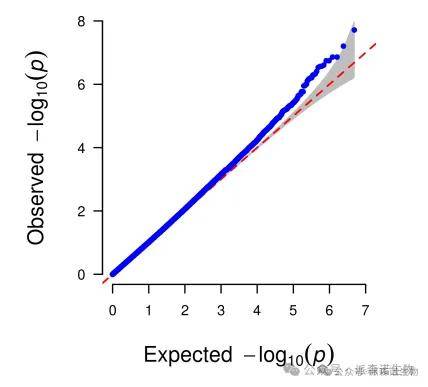
全基因组关联分析
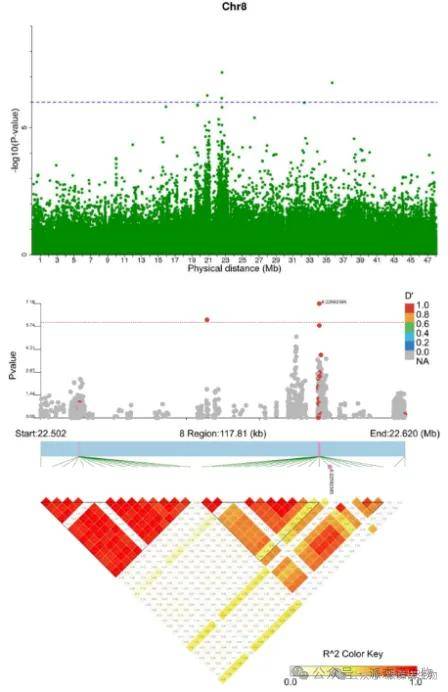
LDBlock联合分析及候选基因关联分析
● 选择性清除分析(高级分析)
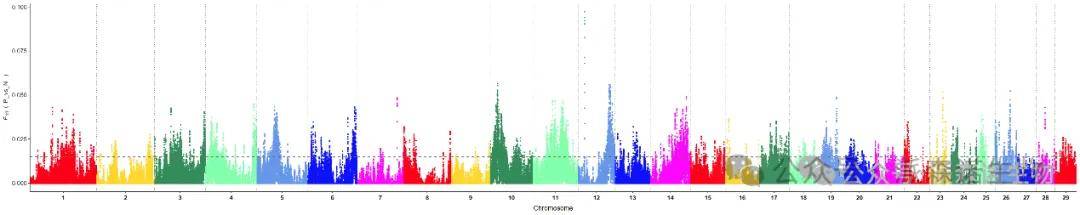
选择性清除分析
精选案例
文章题目:Genome- and transcriptome-wide association studies provide insights into the genetic basis of natural variation of seed oil content in Brassica napus
研究材料:505 份甘蓝型油菜自然品种,包括半冬季型和春季型
测序方案:自然群体重测序(平均测序深度~9X)、转录组测序
科学问题:培育高含油量甘蓝型油菜品种具有重要的经济意义,本研究拟通过基因组和转录组关联研究揭示甘蓝型油菜种子油分自然变异的遗传基础。
研究思路:
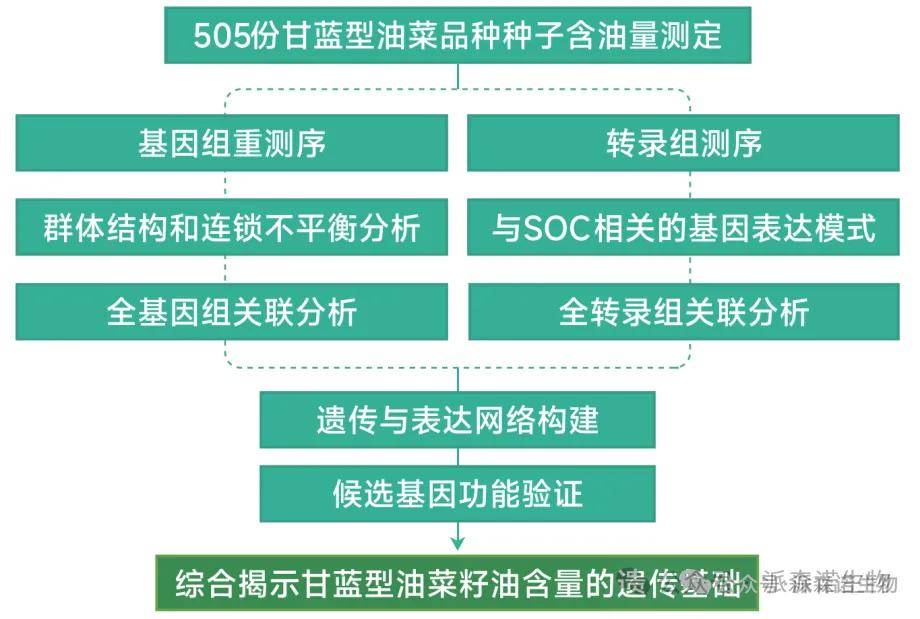
核心内容展示:
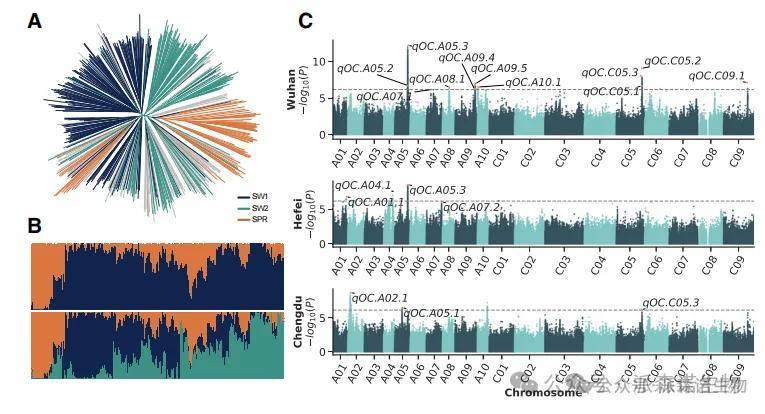
甘蓝型油菜自然群体种子含油量GWAS分析
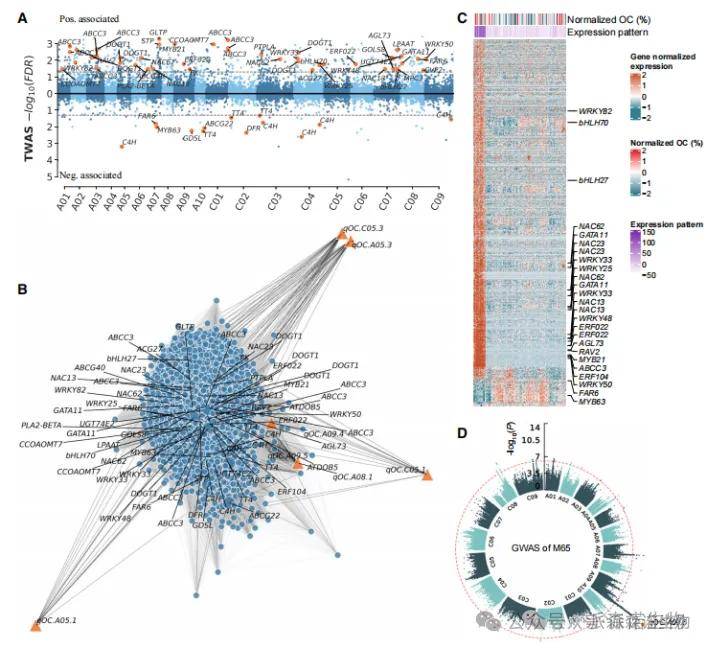
甘蓝型油菜自然群体种子含油量TWAS分析
参考文献:Tang S, Zhao H, Lu S, et al. Genome- and transcriptome-wide association studies provide insights into the genetic basis of natural variation of seed oil content in Brassica napus. Mol Plant. 2021, 14: 470-487.


















