

2025-06-18

随着转录组、蛋白组、代谢组甚至表观组技术的不断发展,海量组学数据为我们揭示重大生物学现象提供了空前机会。如何从如此庞大的数据中稳定地挖掘出具有重大生物学价值的标志物(Biomarkers),成为现代生物学和转化医学关注的一大挑战。
那么,有哪些方法可以帮助我们从数据中甄别出有价值的Biomarkers呢?下面以派森诺基因云宏基因组云分析为例,一起看看发现Biomarkers的主要方法。
1.从多元统计图上选择
PCA(主成分分析)的载荷图可以帮助我们直观地甄别对样本分离贡献大的分子。降维时,会求出每一个变量(如不同物种)的“loading”数值,衡量它对每一个主要成分(PC1、PC2)的贡献大小,取值范围为-1到1。
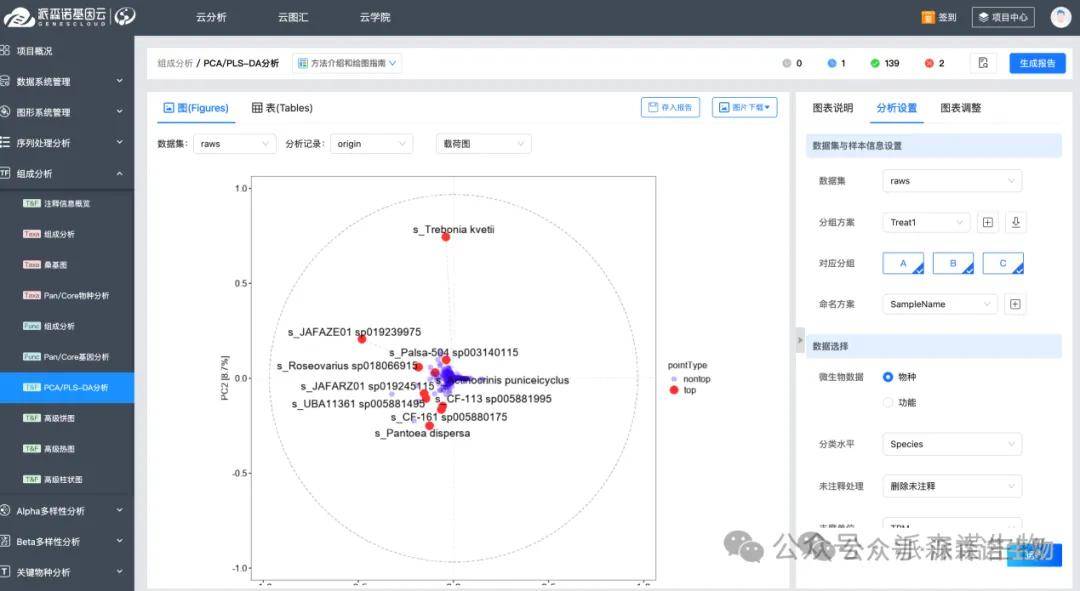
2.通过差异分析结果选择
我们可以对不同组之间进行统计学差异检测(如 Student’s t-test 或 Wilcoxon rank-sum test),或者借助 ALDEx2、ANCOM 等软件进行比较,获取每种分子的 p 值和 FDR 值。由此可以初步锁定出差异显著(p 候 < 0.05,FDR < 0.05)的标志物。
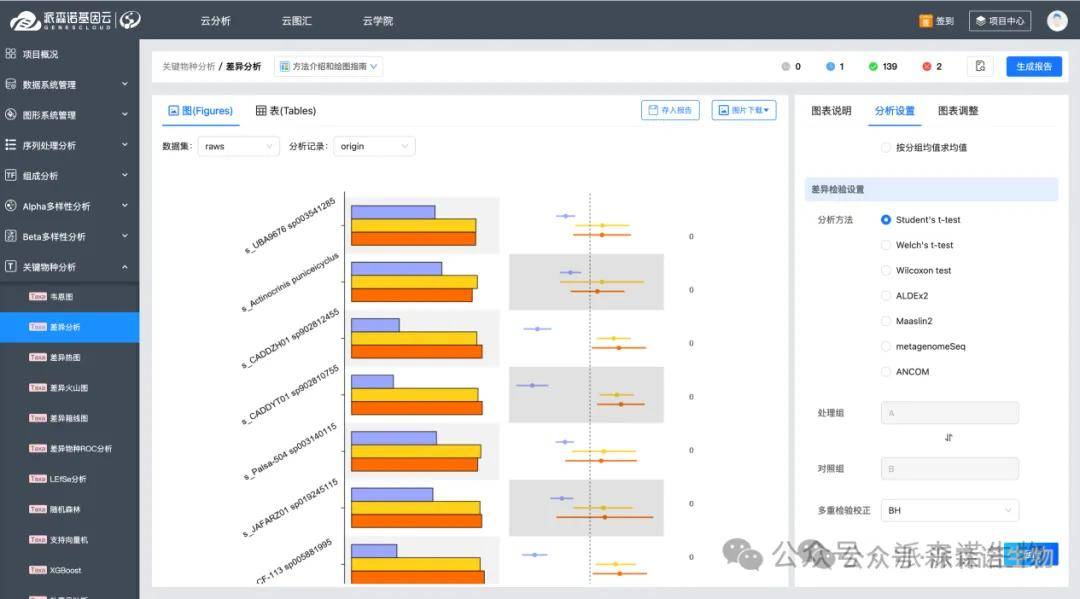
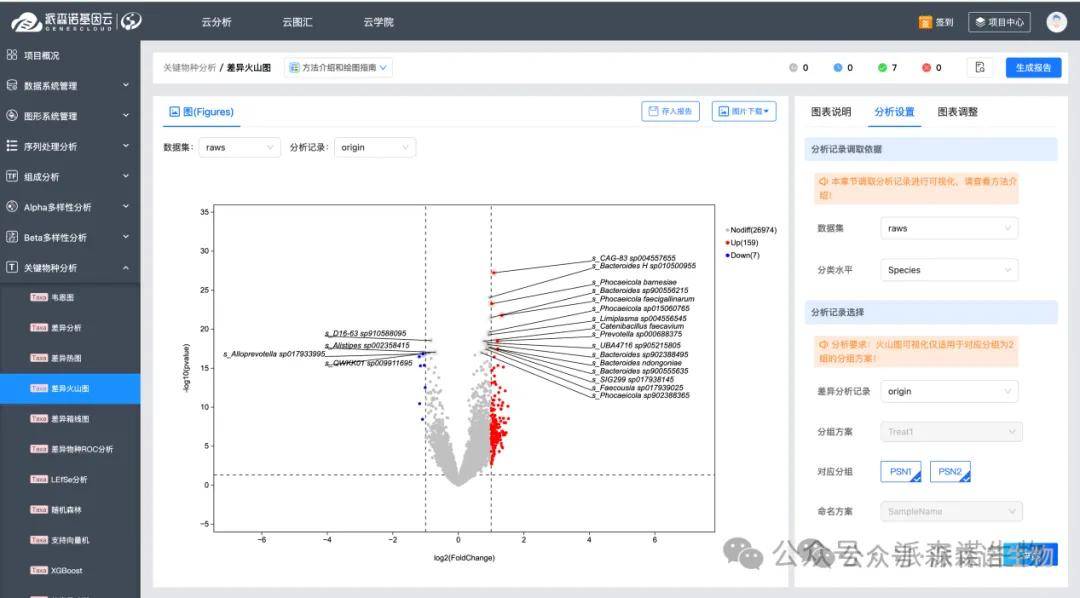
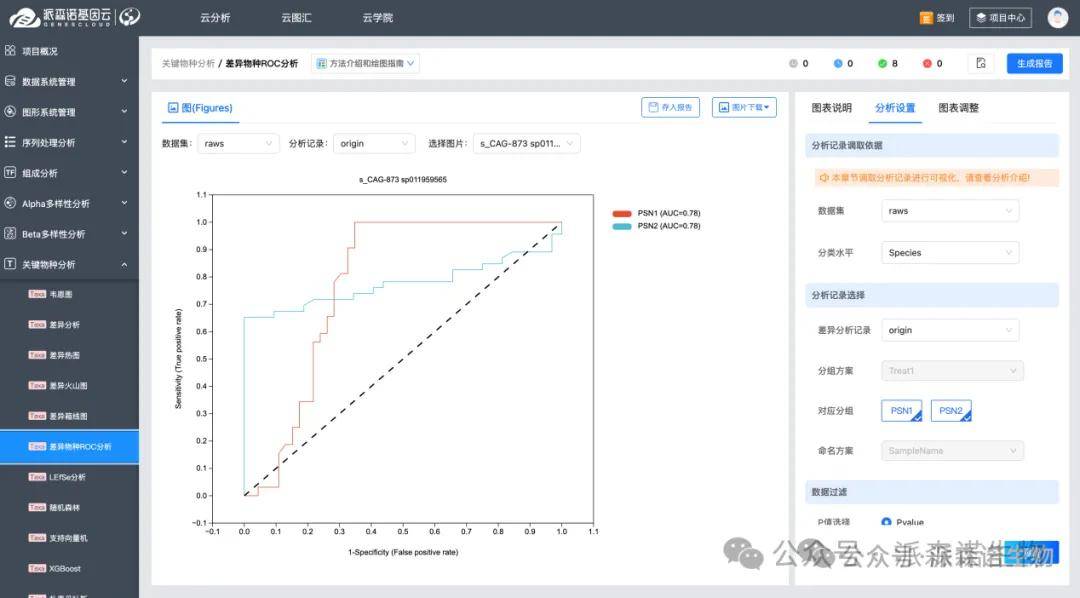
3.通过ROC曲线分析候选biomarker的诊断价值
我们可以对上述差异分子进行ROC 曲线(受试者工作特性曲线)分析。具体来说,以生物标志物的丰度为预测变量,样本分组为标签,计算不同阈值下的真阳性率(TPR)和假阳性率(FPR),最终形成 ROC 曲线。若 AUC 值接近 1,表明标志物的诊断能力越好。
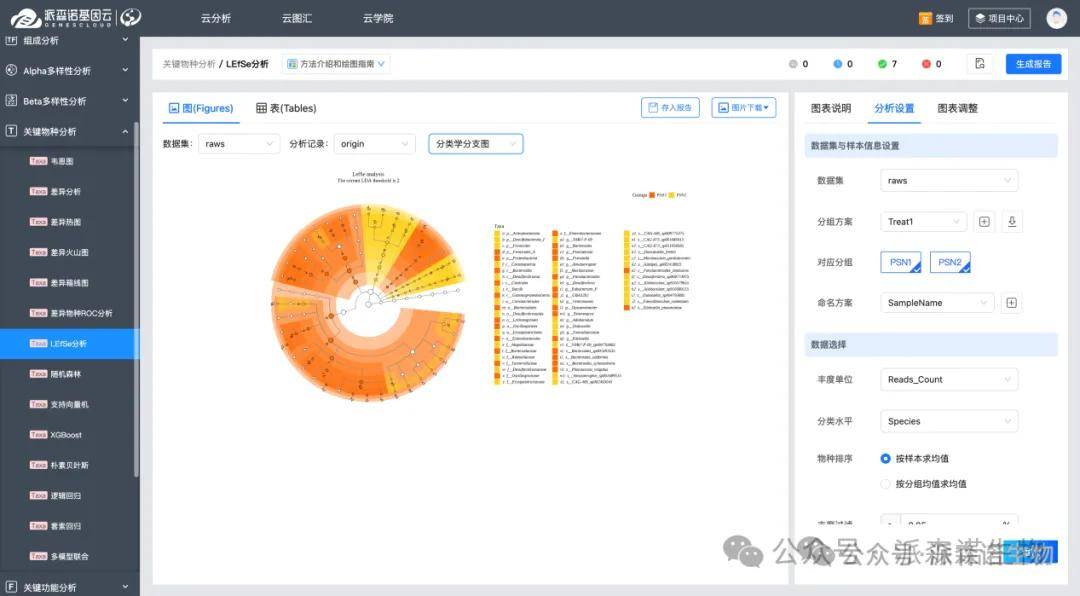
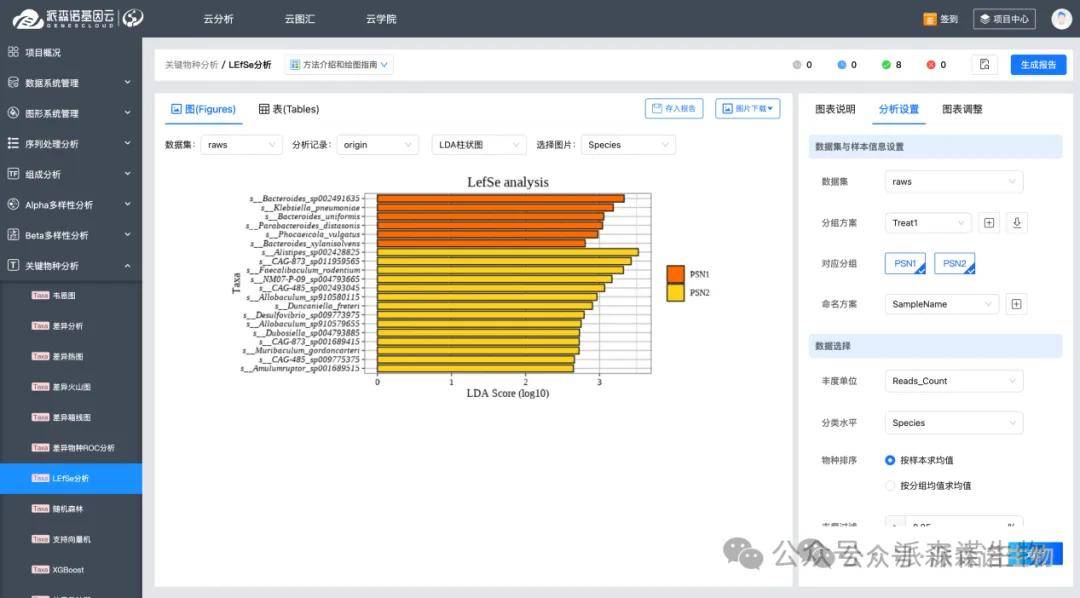
4.通过LEfSe分析LDA效应大小选择biomarker
我们可以通过LEfSe(Linear Discriminant Analysis Effect Size)分析不同组之间丰度有显著差异的物种,同时计算出每种物种对分组差异的贡献大小(LDA 值)。一般来说,LDA 值越大,表明该物种对分组差异的作用越重要。我们通常设置一个 LDA 阈值(如 2 或 4),大于该阈值的物种就可以被认为是具有生物标志物价值的候选。
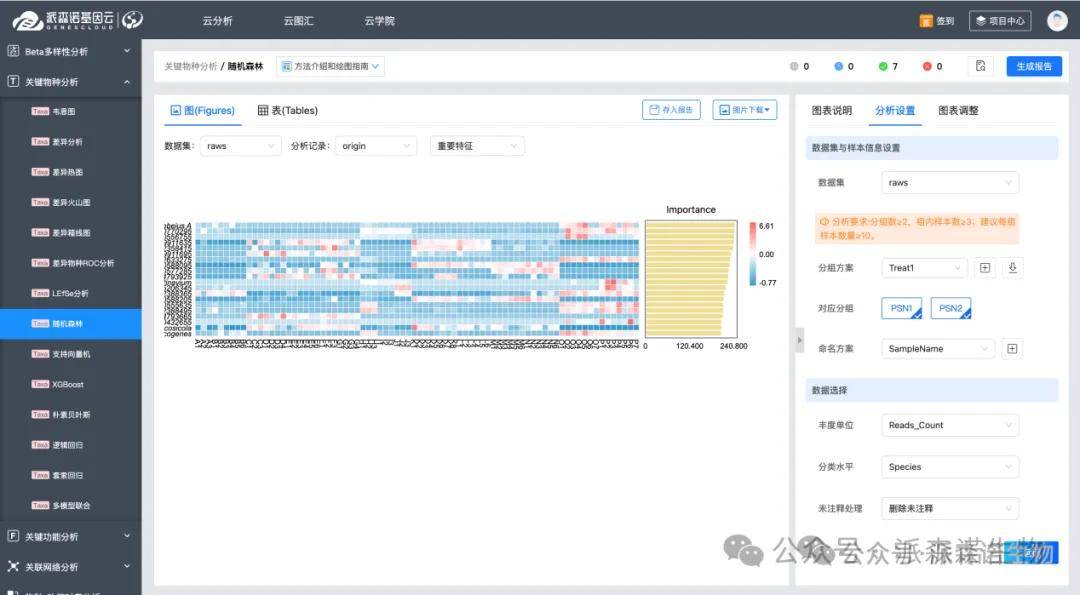
5.机器学习建模进行标志物甄选
我们可以借助 Random Forest、LASSO 或 Support Vector Machine 等机器学习模型进行建模,一方面可以衡量模型对样本标签的预测准确性,另一方面可以获取每种分子对模型预测所起作用的重要性分数,从而甄别出关键标志物。
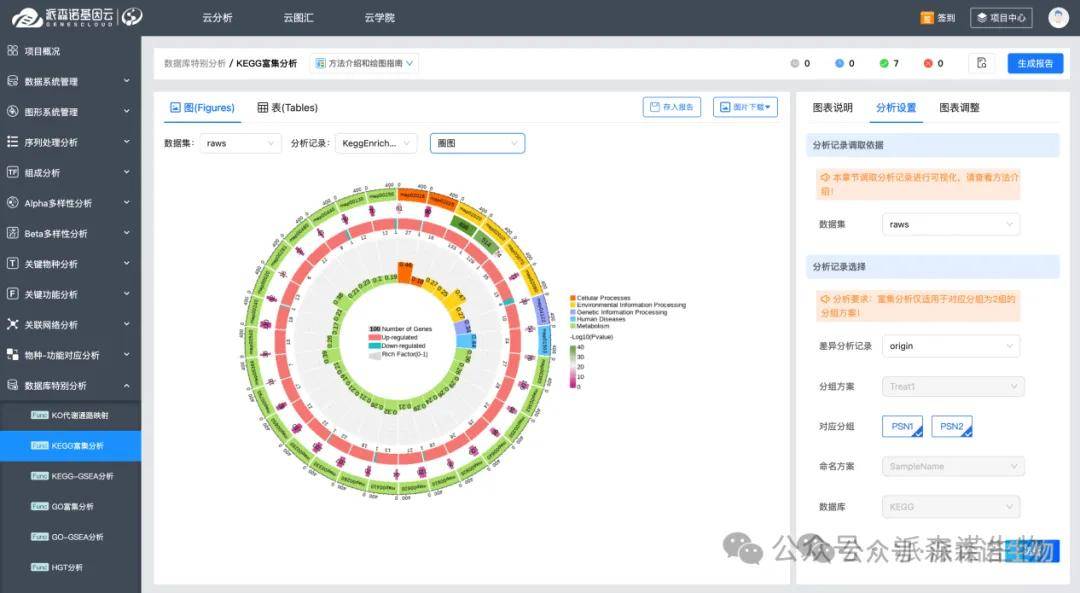
6.通过功能富集锁定通路
使用富集分析从基因变化看从整体功能层次变化,揭示它们主要富集于哪个可能与生理功能高度相关的生物过程和信号路径。常见的富集分析方法有基于差异基因的超几何检验分析和基于全部基因的GSEA分析方法。富集分析常用于宏基因组、转录组学等分析中。
7.相关性分析
使用相关性网络分析单组学数据内部共现情况或两组学、三组学间信息关联度。网络分析中高度连通(hub)的节点可以被认为是在整体网络中起到“关键调控作用”的分子,因而它们是值得优先关注的biomarker。此外,与关键表型信息高度相关的分子标记也可以作为候选biomarker。
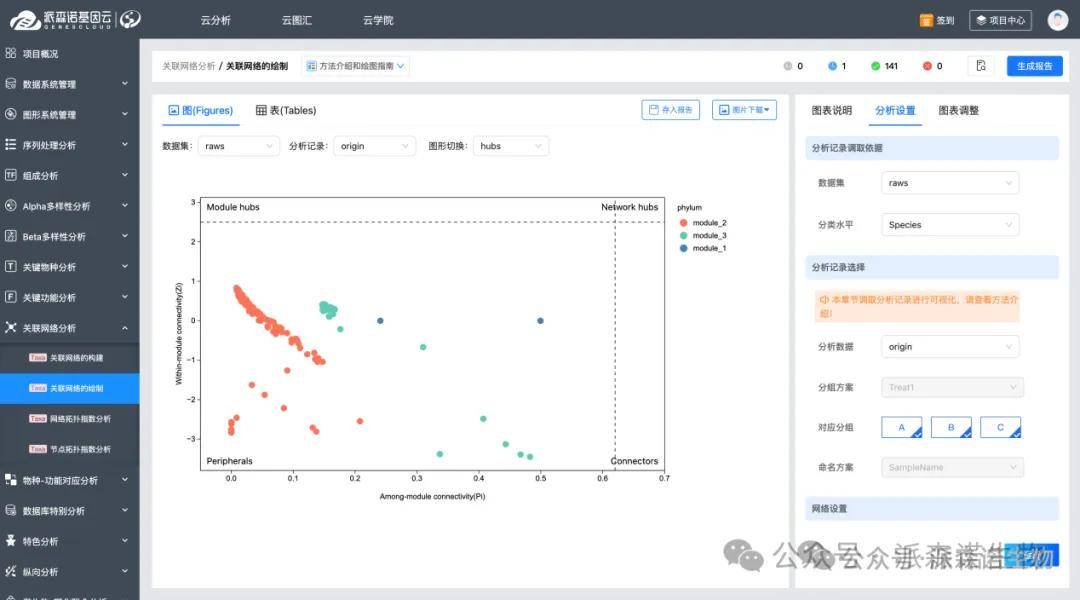
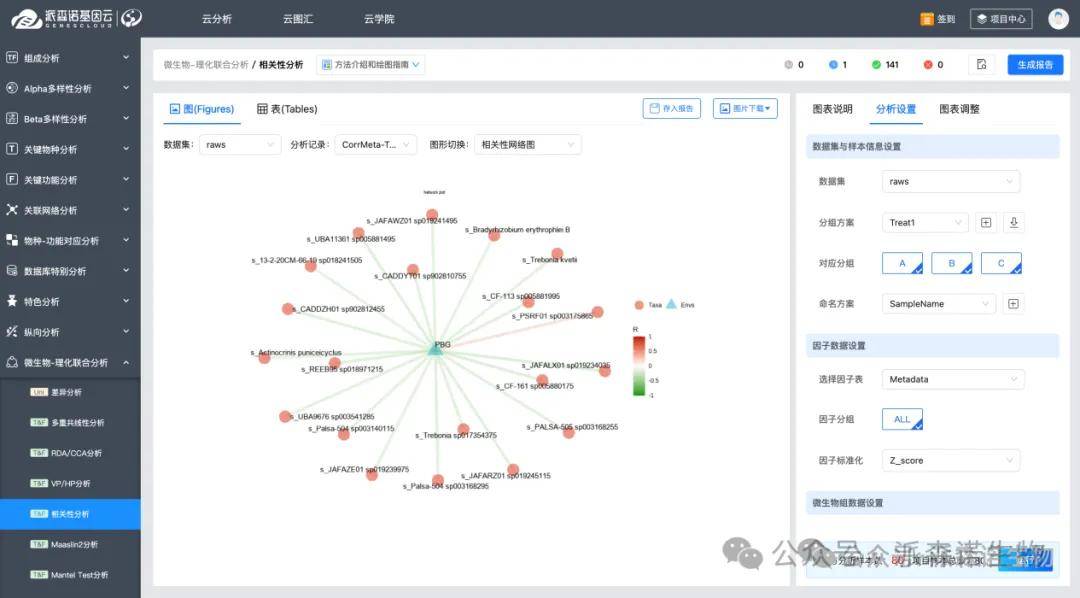
最后,基于组学数据挖掘的biomarker,往往需要通过验证阶段:
(1)同实验材料验证/同队列验证:
通过qPCR、Western blot 或靶向质谱对所挑出的标记进行实验验证,确保它们在不同样本中均表现稳定且具有良好重复性。
(2)跨实验材料/跨队列验证:
从训练集到验证集,再到外部数据或不同物种的验证,是标志物筛选的“黄金标准”。
案例分享
大队列血浆蛋白质组揭示脓毒症亚型和生物标志物
Mi, Y. et al. High-throughput mass spectrometry maps the sepsis plasma proteome and differences in patient response. Sci. Transl. Med. 16, eadh0185 (2024).
脓毒症(Sepsis)是一种由感染导致的全身性炎症综合征,伴有器官功能障碍,严重时甚至发生败血性休克和多器官功能衰竭。为了寻找可以辅助诊断和指导治疗的新标志物,本文对1611 名患者(共 2612 个血浆样本)的蛋白质组进行大规模检测。通过PCA分析发现,PC1的高正载荷蛋白包括急性期反应蛋白、S100类促炎蛋白、先天免疫或抗菌蛋白以及细胞外基质(ECM)蛋白等,而脂质转运蛋白APOM则表现出高负载荷,表明血浆蛋白质组的变化与疾病的严重程度密切相关。研究进一步通过差异分析、富集分析、蛋白丰度与疾病严重程度的相关性、ROC分析等,综合锁定了潜在的生物标志物,为脓毒症的精确医学方法提供了方向。
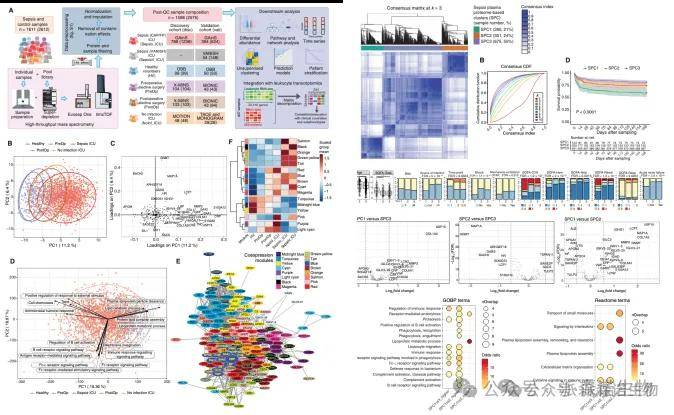
研究部分结果展示


















