

2017-03-01
以PacBio公司的SMRT单分子实时测序技术(Single molecule real-time sequencing)为代表的三代测序技术,通过其独有的环形一致性测序模式(Circular-consensus sequence,CCS),极大提高单碱基测序的准确率,远超Illumina等二代测序技术。与传统转录组测序项目相比,利用PacBio平台的全长转录组测序技术可以直接获得mRNA的全长,保证了mRNA序列的精确性。近期我们将陆续推出全长转录组测序技术相关文章,供讨论和交流。我们在上一期向大家介绍了全长转录组测序的技术原理和实验流程,本期将为大家介绍全长转录组测序分析流程中的数据整理和质控。
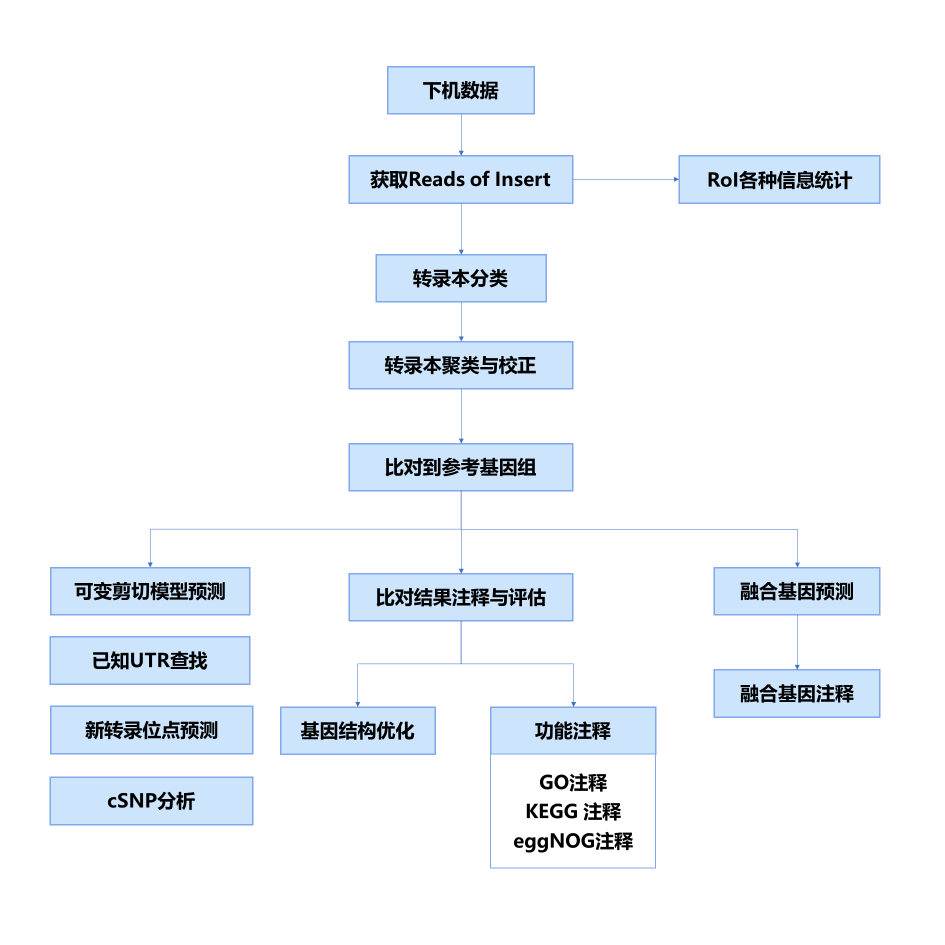
分析流程
首先从下机数据中提取Reads of Insert(RoI)序列,根据Reads of Insert序列是否含3’ 端引物和5’端引物以及是否嵌合对Reads of Insert进行分类,对全长序列进行去冗余聚类,并用非全长序列对其进行校正。接着将非冗余序列比对到该物种的参考基因组上,在此基础上完成融合基因分析,并进行比对结果整理,根据比对结果与参考基因组的注释信息对Isoform进行功能注释,并完成基因结构优化。另外,我们还进行了转录本结构分析,包括可变剪接分析, UTR区域注释,cSNP 和InDel 分析等。
数据整理和质控
每个原始序列可以分割成一个或多个子序列(Subread,Subread 是DNA聚合酶以一条模板链经过一个Passes合成的,不包括Adapter序列),即每个零模波导孔中会有多个Subreads。每个零模波导孔中的所有Subreads来自同一个转录本,由于其碱基出错率是随机的,可通过Subreads间比对提高碱基质量,获得一条Reads of Insert,即Reads of Insert通过同一零模波导孔中的Subreads校正后得到。
对原始下机数据进行提取和过滤Subreads,去除Adapter和低质量的序列。为了充分利用数据,筛选出长度大于50bp、序列准确度大于0.8并且Full Passes数目大于0的序列,得到Reads of Insert,如图1。一个Full passes指原始序列中的一条子序列两端均含有 Adapter(图中黑色区域),一个原始序列的Full passes数目指在该序列中文库cDNA序列被完整测到的次数,图1中有Full passes数目为2。
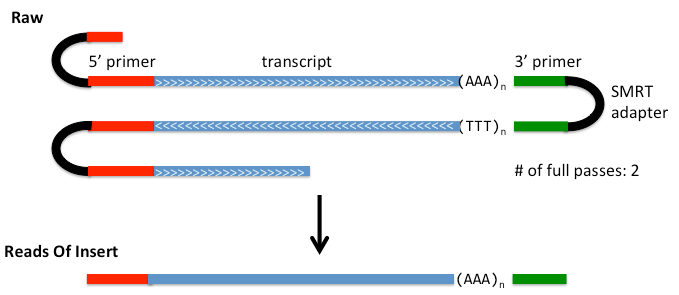
图1 Reads of Insert示意图
我们对每个样品的Reads of Insert按不同插入片段长度分别进行统计,包括RoI序列数目、RoI总碱基量、RoI序列平均长度、RoI序列平均质量和平均Passes。整理好的数据就可以开展后续的分析了,具体分析内容将在下一期进行阐述,敬请期待。


















