

2025-07-30

文章信息
英文题目:Evolutionary origin of genomic structural variations in domestic yaks
中文题目:家牦牛基因组结构变异的进化起源
发表期刊: nature communications
影响因子:15.7/Q1
发表时间:2023.09.19
涉及组学:基因组组装、泛基因组构建、SV结构变异、选择性清除分析、SNP&SV-GWAS。
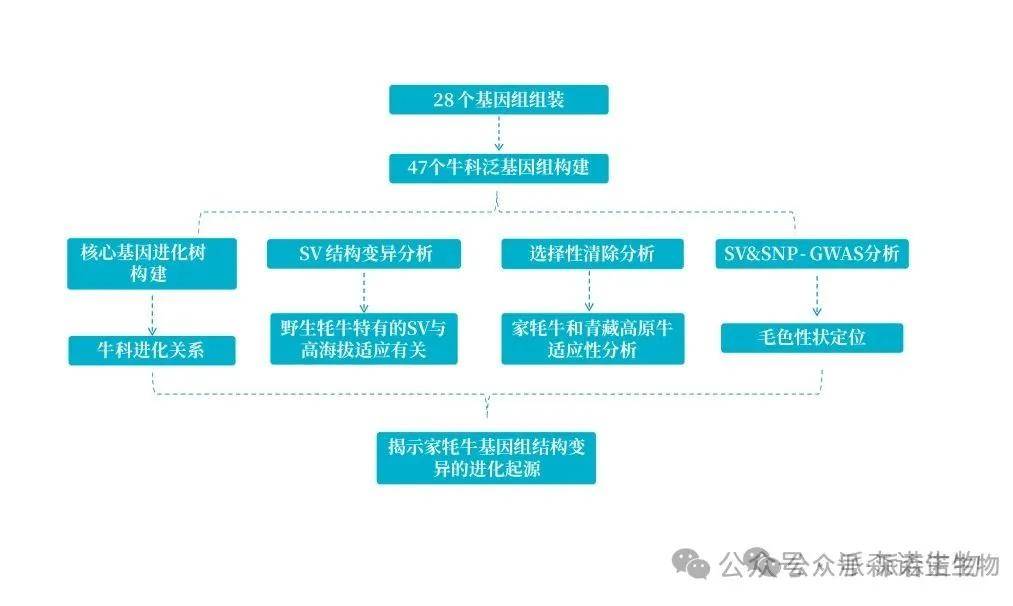
技术路线
摘 要
牦牛在进化过程中经历了自然选择、人类驯化和种间基因渗入。然而,这些过程各自青睐的遗传变异此前尚未被明确区分。研究人员构建了7种可杂交牛科物种的47个基因组图谱,通过386个个体的基因分型,成功检测到57,432个高分辨率结构变异(SVs)在物种内部和跨物种间的分布。通过系统发育分析,研究人员明确了家养牦牛中各类SVs的进化起源。进一步发现334个与家养牦牛SVs重叠的基因可能携带来自野生牦牛的选择信号,另有686个基因存在牛源渗入。近90%的家养牦牛存在牛源渗入现象。跨越KIT基因的SV渗入触发了白色家养牦牛的培育。研究人员验证了筛选出的分层SVs与基因表达存在显著关联,这有助于解释表型变异。研究结果表明,不同来源的SVs共同促进了家养牦牛的表型多样性。
前 言
牦牛是牛科动物,包括野生种(Bos mutus,约1.5万头)和家养种(Bos grunniens,约1800万头)。野生牦牛在500万年前就适应了青藏高原4000-6000米的高海拔极端环境,而家牦牛主要生活在3000-5000米区域,为人类提供资源。家牦牛在进化中经历了自然选择、人工驯化以及与牛类的基因渗入,成为研究遗传进化的理想模型。
研究发现,基因组结构变异(SVs)在牦牛的高海拔适应和驯化中起关键作用。通过构建泛基因组和超级泛基因组,研究人员分析了28个新组装的牦牛和亚洲牛基因组,以及19个已发表的近缘物种基因组,并在386个个体中鉴定出SVs。这些变异影响基因表达,促进缺氧适应、驯化及种间基因渗入,深化了对牦牛基因组多样性和适应性遗传机制的理解。
研究内容
1.野生牦牛和家养牦牛的系统发育关系和泛基因组
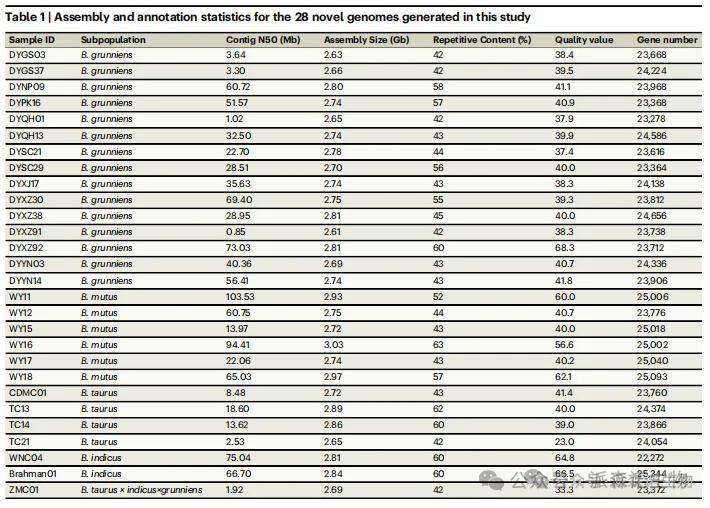
为解析野生牦牛与家养牦牛的基因组多样性,研究人员构建了6头野生牦牛、15头家养牦牛(覆盖其完整分布范围)、2头低海拔亚洲水牛、4头高海拔塔乌牛,以及1头高海拔塔乌-水牛-牦牛杂交种(张牧牛,ZMC)的从头全基因组测序数据(表1)。这些数据与已报道的1头野生牦牛、14头牛、2头野牛、1头欧洲野牛和1头印度野牛基因组数据整合后,形成了牛科动物基因组数据集(表1)。为确保分析一致性,研究人员采用统一标准流程对这47个牛科基因组进行注释(表1)。每个组装平均鉴定出24,368个蛋白质编码基因(表1),通过BUSCO评估显示平均97. 4%的蛋白质编码注释完成度,表明基因注释高度完整。
在这些基因组组装中检测到1,048,639个高置信度SNP,并以水牛基因组为外群进行系统发育分析(图1a)。研究人员基于每个物种选取一个代表性个体,通过8,428个单拷贝核心基因构建了物种树(图1b)。两棵系统发育树与既往研究结果高度吻合3,4,但家养牦牛未形成单系聚类(图1a)。研究人员构建了牦牛和牛的泛基因组,并为7个牛科物种构建了超级泛基因组(图1c)。牦牛泛基因组在n = 20时总基因集接近饱和(图1c)。核心基因家族(存在于全部22个基因组)、近核心基因家族(存在于20-21个基因组)和可变基因家族(存在于1-19个基因组)的占比分别为50.18%、10.91%和38.91%(图1d),其中核心与近核心基因表现出更高的平均表达水平和更低的Ka/Ks比值(图1e,f)。牛的泛基因组及7个牛科物种47个基因组的超级泛基因组也呈现类似趋势。平均而言,家养牦牛基因组包含119个野生牦牛基因组中缺失的基因,其中56个通过基因渗入源自牛(图1g),这些基因在疾病防御、发育和繁殖功能方面具有显著富集特征。在构建泛基因组的基因组对比较中,每个基因组均包含123至2113个其他基因组缺失的基因(图1h)。
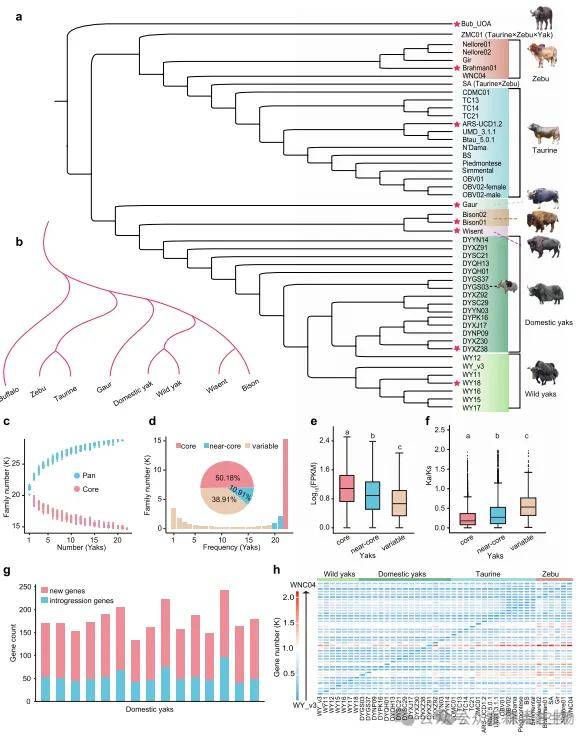
图|牛科动物系统发育及野生与家养牦牛泛基因组
2.牛图形基因组和7个物种中386个个体的结构变异(SV)的特征
为识别结构变异(SVs),研究人员基于47个基因组构建了多组装图谱基因组,包含3.14 Gb序列,分布在5,449,222个节点和4,889,530条连接边上,其中非参考节点覆盖387.0 Mb(图2a、b)。节点分为核心(所有基因组共有,占60.8%)、近核(存在于45-46个基因组,占17.0%)和可变(存在于≤44个基因组,占22.2%)。使用gfatools检测到293,712个可基因分型的SVs(81.7% <500 bp,99.76% <10 kb),其中40.8%为多等位型(覆盖76 Mb),在重复DNA区域富集。33.1%的SVs位于潜在表达调控区或编码序列(98.2%在调控区,1.8%在CDS),12.97%的多等位型SVs影响了54.4%的参考基因。对386个样本(覆盖深度>6×)进行分型,获得57,432个高质量SVs(召回率0.96)。遗传分析显示,多数SVs呈物种特异性分布,但牦牛与肉牛存在单倍型交换。30.6%的SVs与邻近SNP存在强连锁不平衡(LD R²≥0.45)。
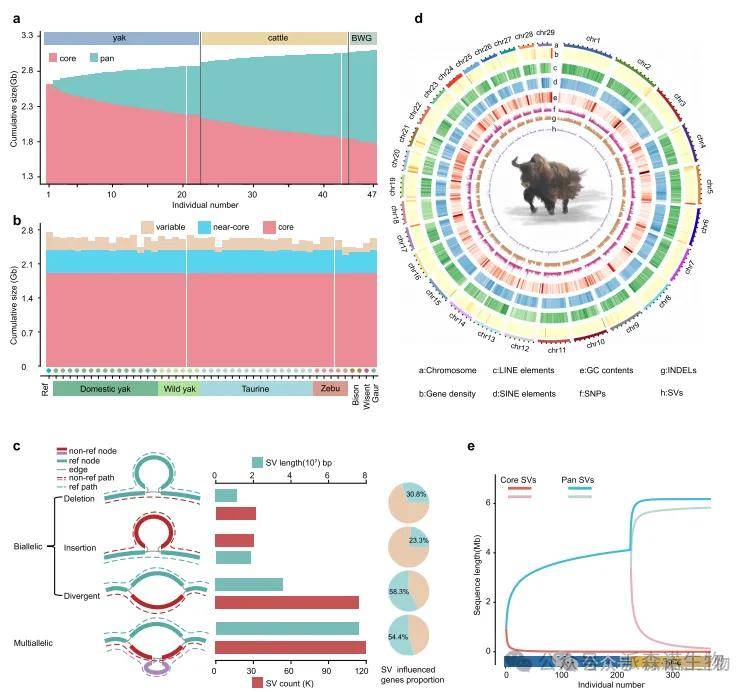
图2|47个新生牛基因组的图谱基因组特征及其结构变异(SVs)在这些基因组和386个个体中的分布
3.野生牦牛特有的sv与高海拔适应有关
研究人员通过计算野牦牛与低海拔牛科动物(野牛、欧洲野牛和欧洲水牛)的固定指数(FST),识别出4830个可能经历选择性清除的结构变异,涉及1051个基因,这些基因在缺氧反应、血管生成等HIF-1信号通路中显著富集(图3a、b)。其中,EPAS1基因内含子的254 bp插入片段(与LINE1元件重叠)在野生牦牛中普遍存在,但家养牦牛频率较低(图3c、g)。该变异降低了启动子活性,导致野生牦牛EPAS1表达水平低于牛类(图3e)。此外,MB基因的155 bp缺失(MB-hap-1)增强了增强子活性(图3d、f、h),可能与牦牛缺氧适应相关。其他候选基因(如PPARA、BCL2、EGFR、IGF1R和IL6RA)也发现重要变异,其功能有待进一步研究。
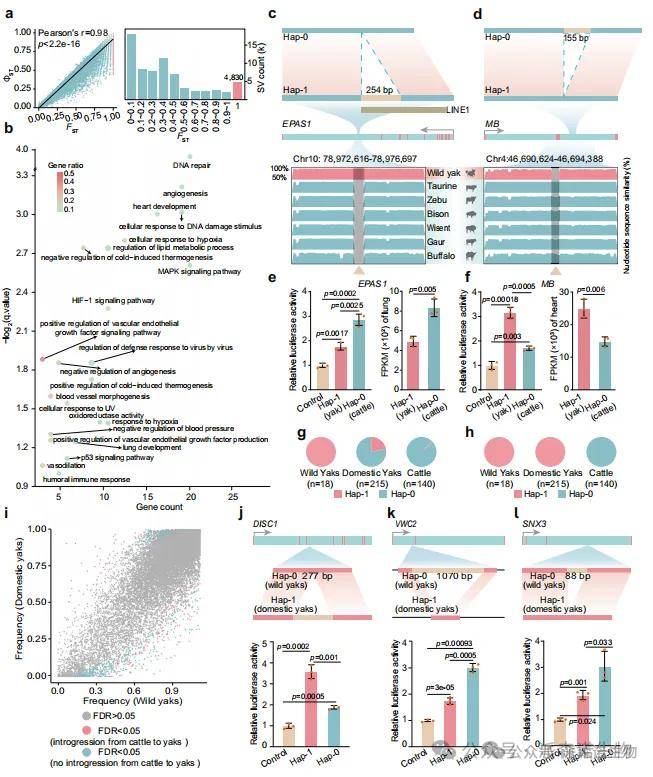
图3 | SVs促进了牦牛的高海拔适应和驯化。
4.家牦牛中SVs的来源及白牦牛的来源
为解析家牦牛结构变异(SVs)的起源,研究人员对30头家牦牛和青藏高原牛(QTP牛)进行系统发育分析,发现26,591个参与种间基因交流的SVs(占总SV的46.3%)。其中91.5%与转座子元件重叠,且牛源SV渗入呈现自东向西递减趋势。值得注意的是,11.6%的牦牛(主要分布在青藏高原北部)未携带牛源SV单倍型。
基因渗入分析显示:
11,486个SV(20.0%)与1151个牦牛向QTP牛渗入的高海拔适应基因重叠;
8,557个SV(14.9%)参与牛向牦牛的基因渗入,涉及抗病性和发育相关基因;
6,547个SV(11.4%)呈现双向渗入模式,如EPAS1基因在高低海拔种群间的互渗。
毛色性状研究发现:
KIT基因的串联易位等位基因(Cs6和Cs29)从彩色侧牛渗入牦牛,导致白色毛色表型;
Hi-C分析揭示白牦牛的KIT基因区呈现抑制性染色质状态(B区室),而黑牦牛为活性态(A区室);
转录组和免疫组化证实白牦牛KIT表达显著降低,与其黑色素缺失表型一致。
这些发现揭示了SVs通过改变染色质结构和基因表达,在牦牛适应性进化和表型变异中的关键作用。
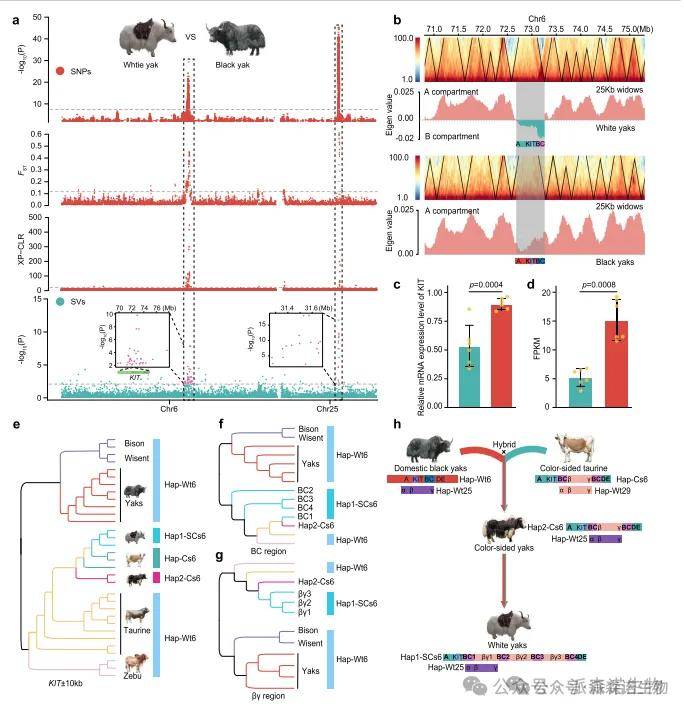
图4|带有从牛体内导入的KIT基因的串联易位SV归功于牦牛的白色毛发颜色。
总 结
研究人员整合28个新测序基因组和已发表数据,构建了牦牛-牛泛基因组及7种牛科动物的超级泛基因组。基于386个样本的分析揭示了重要进化变异,数据可通过牛科泛基因组数据库(http://bovpan.lzu.edu.cn)获取。需注意的是,数据主要来自青藏高原牦牛和中国牛群,不同测序技术(如ONT和PacBio HiFi)导致重复区域覆盖度存在差异。尽管如此,研究仍鉴定出多个与高海拔适应相关的SV,区分了家养特征和牛类基因渗入。例如,发现EPAS1、MB等新候选基因,其中EPAS1存在双向渗入现象。此外,证实KIT基因易位SV导致牦牛毛色变异,且近90%家牦牛携带牛源杂交成分。这些发现对指导家牦牛育种具有重要意义。
参考文献:Liu, X., Liu, W., Lenstra, J.A. et al. Evolutionary origin of genomic structural variations in domestic yaks. Nat Commun 14, 5617 (2023). https://doi.org/10.1038/s41467-023-41220-x


















