

2025-08-10

文章亮点
1.文章完成65个不同人群、130条近乎完整(T2T)的单倍型基因组组装,显著提升了结构变异检测精度,填补了着丝粒、MHC、SMN1/2等复杂区域的空白。
2.组装并分析1,246个人类着丝粒,发现α卫星阵列长度差异达30倍,7%的染色体存在“双动粒”结构,并系统揭示Alu/L1等移动元件在着丝粒的插入模式。
3.派森诺提供泛基因组分析一站式解决方案:从样本检测到基因组分析,专业解析基因组的复杂差异和变化机制,助力生物学和医学研究突破。
文章信息
文章题目:Complex genetic variation in nearly complete human genomes
中文题目:近乎完整人类基因组中的复杂遗传变异
发表期刊:Nature
影响因子:48.5
发表时间:2025年7月
涉及组学:基因组Denovo、泛基因组分析、转录组测序
技术路线
摘 要
本研究对65个具有多样性的人类基因组进行测序,构建了130个高质量的单倍型组装,不仅闭合了92%的组装缺口,还使39%的染色体实现了端粒到端粒的完整组装,全面解析了复杂区域如MHC、SMN1/SMN2等和1,852个结构变异。同时,首次完整组装并验证了1,246个着丝粒,揭示其在α-卫星重复序列长度和移动元件插入方面存在高度变异。结合现有泛基因组参考,该成果显著提升了短读长数据的基因分型准确性,每个个体可检测到约26,000个结构变异,为人类基因组多样性研究和疾病关联分析提供了关键资源。
研究背景
长读长测序(Long-read sequencing, LRS)技术在完成首个人类基因组时发挥了关键作用。LRS显著提升了结构变异(SVs,定义为长度≥50 bp的变异)的检测灵敏度。本研究基于人类基因组结构变异联盟(HGSVC)的最新工作,利用PacBio HiFi与Oxford Nanopore超长读长等互补的长读长测序技术,结合Hi-C、Strand-seq和三亲本信息,在65个来自千人基因组计划的多样性人群中,构建几乎无缺口的高质量染色体组装,重点解决了以往在着丝粒和高同源段复制区域中存在的组装难题,为全面解析人类结构变异和完善泛基因组参考奠定了基础。
研究结果
1.130个单倍型基因组组装
研究人员选取来自五大洲28个人群的65位个体,利用PacBio HiFi与Oxford Nanopore超长读长测序技术,结合Strand-seq、Hi-C等辅助数据,构建了130个高质量、单倍型解析的人类基因组组装。使用Verkko工具,实现了中位数为10条染色体的端粒到端粒(T2T)组装,以及中位数为8条染色体的高质量scaffold组装,显著提升了组装连续性和准确性,单拷贝基因的组装完整性达99%。针对T2T-CHM13参考基因组,检测到约18.8万个结构变异、630万个indel和2390万个SNV;对GRCh38参考组也获得了相应变异集。相比以往研究,SV检测量提高59%,假阳性率降低55%。
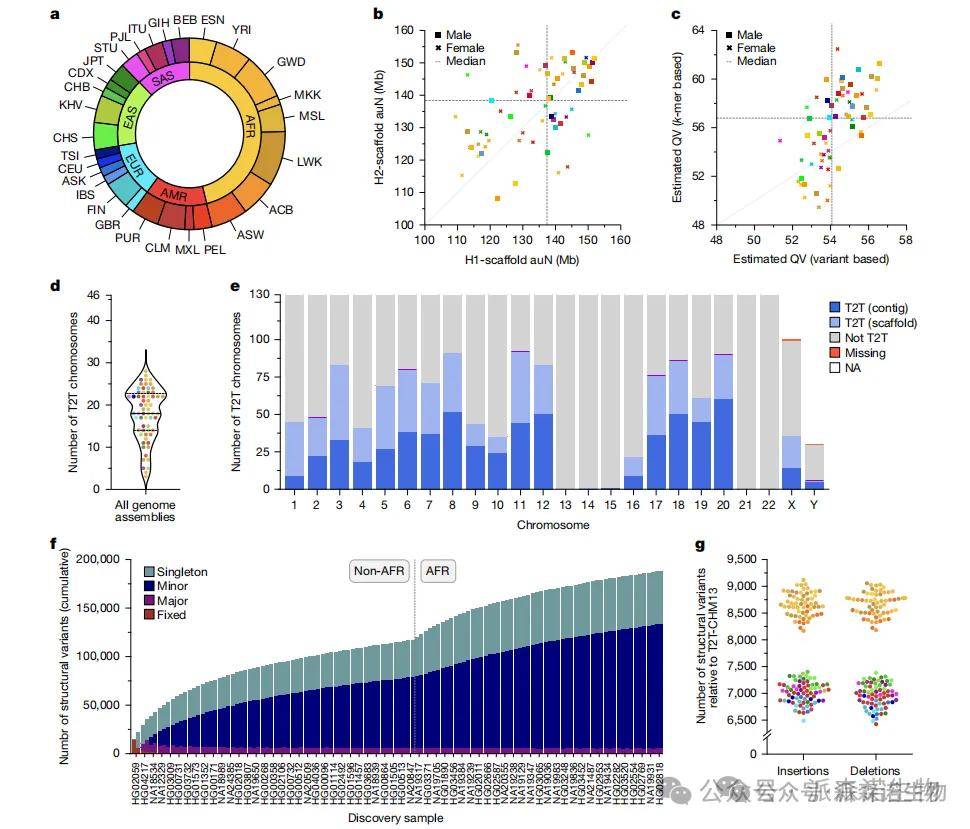
图1 65个人类基因组的组装和变异识别
2.基因组资源挖掘
研究人员系统分析了来自130个单倍型组装的结构变异(SVs),涵盖移动元件插入(MEIs)、倒位、片段重复(SDs)和Y染色体变异等。共鉴定出12,919个MEIs,其中大部分全长L1插入具备潜在反转录转座能力。倒位分析发现多个新倒位事件,包括与Sotos综合征相关的大型倒位。片段重复方面,平均每个基因组含168.1 Mb的SDs,并识别出多个此前未注释的拷贝数多态性区域,尤其在非洲人群中更为丰富。此外,我们高质量组装了30条Y染色体,首次解析了多个Yq12区域的复杂结构。功能层面,1,535个SVs影响了985个蛋白编码基因,其中大多数为多态性变异,部分仍保持基因功能。研究还结合Iso-Seq和Hi-C数据揭示SV对转录本异构性、基因表达及染色质结构的影响。
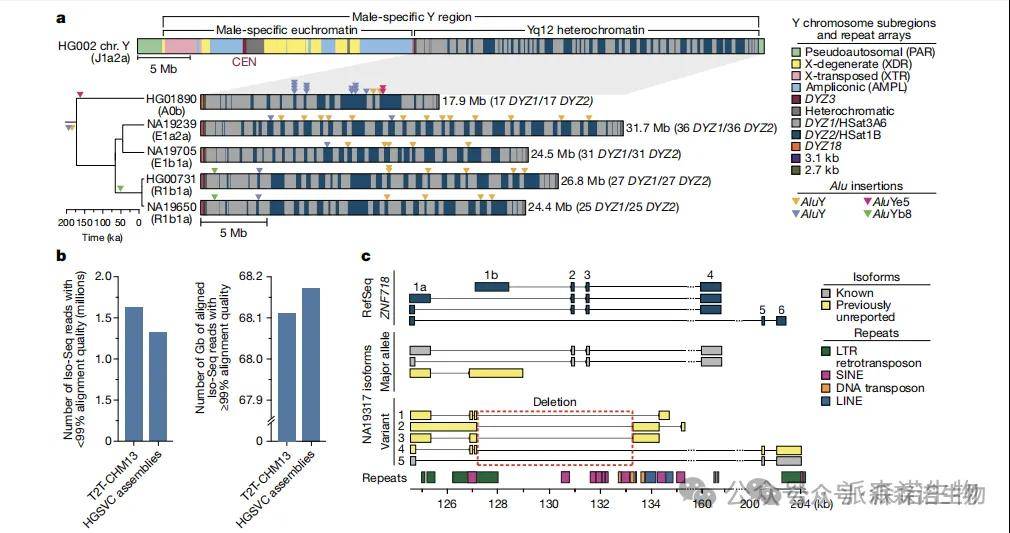
图2 复杂区域改进的基因组资源
3.基因分型综合参考
研究人员通过构建包含107个人类基因组的泛基因组参考图谱,利用PanGenie工具对千人基因组计划(1kGP)3,202个个体进行全基因组变异分型,共检测到超过3,000万个变异位点,包括25.7百万SNP、5.8百万插入缺失和47.9万结构变异(SV)。相比既往HPRC和1kGP-HC数据集,新方法显著提高了稀有SV检出率(非洲个体达1,490个/基因组)。通过整合短读长数据与单倍型定相技术,实现了中位k-mer质量值48的基因组重建,较传统方法显著提升。针对复杂医学相关基因位点,采用Locityper靶向分型使单倍型预测准确率(质量值≥30)从74.6%提升至80%,其中HLA等关键基因分型性能改善尤为显著。研究表明,扩大参考单倍型库可进一步提高难测基因位点的分型精度,为疾病研究提供更可靠的基因组分析工具。
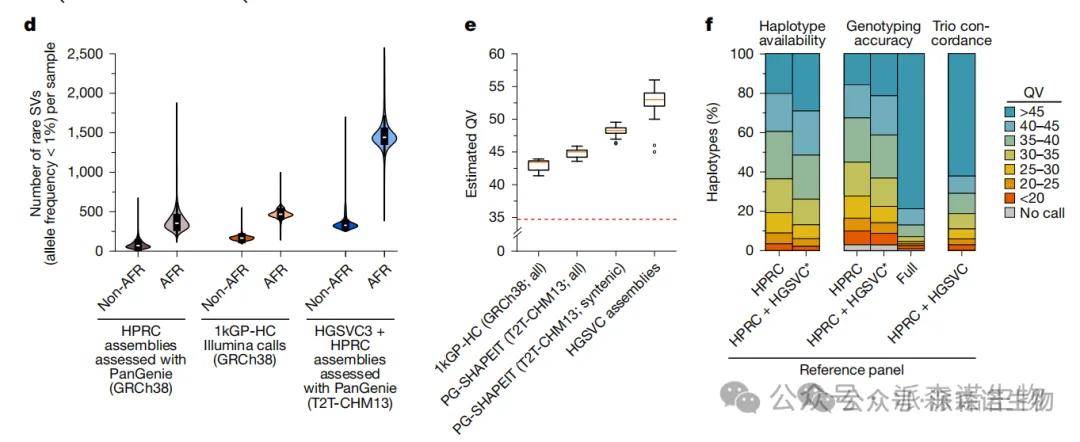
图3 结构变异分型情况比较
4.主要组织相容性复合体(MHC)
研究人员对主要组织相容性复合体(MHC)区域进行了全面分析,注释了130条单倍型中的HLA和非HLA基因,修正了826个不完整的HLA等位基因注释,并发现了170个新的结构变异。研究揭示了MHC II类单倍型(如DR8和DR1)的演化机制,解析了RCCX基因簇的复杂模块结构及其进化中的基因转换事件,同时开发了Locityper工具将等位基因预测准确率提升至97.1%。通过泛基因组分析不仅验证了传统HLA-DR分组系统,还发现了潜在的细分亚类,为疾病关联研究和精准医疗提供了重要资源。
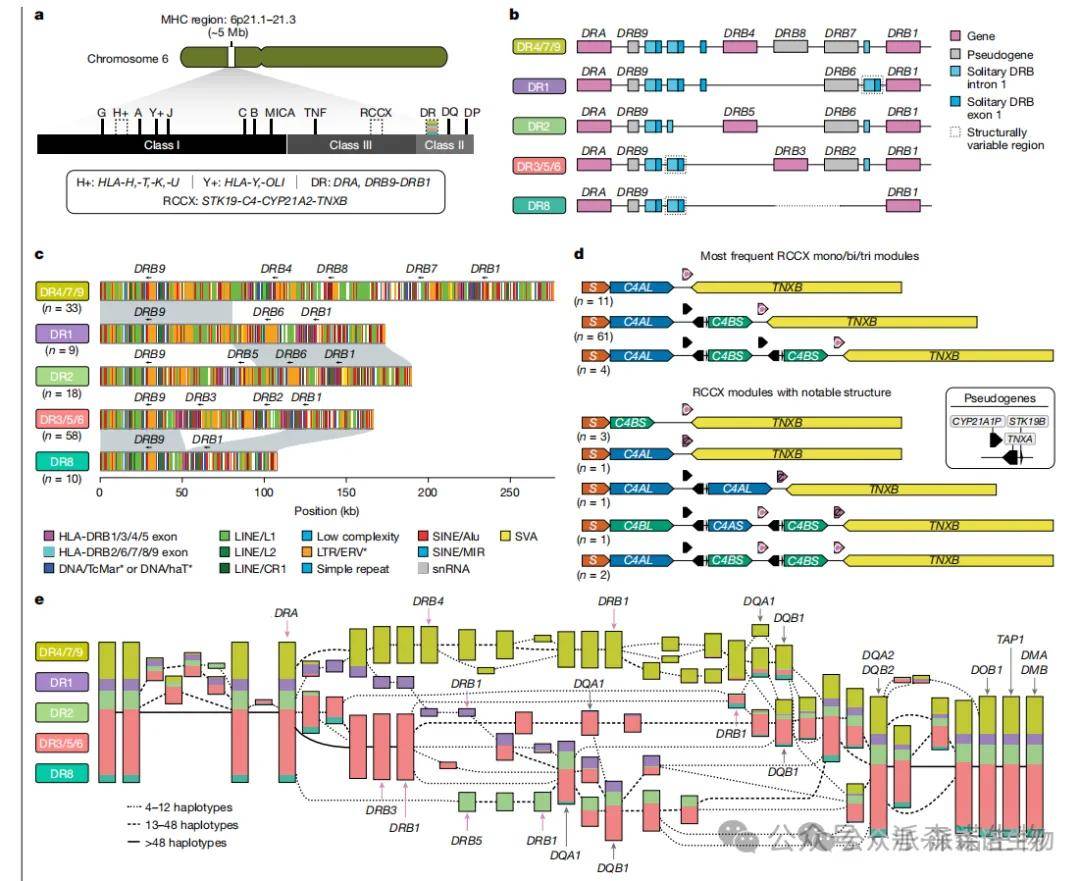
图4 MHC位点的结构可变区
5.SV检测与泛基因组构建
长读长组装基因组显著提升了复杂结构变异(CSV)的检测能力,尤其是在重复序列(如串联重复和转座元件)中更准确地识别变异。最新的PAV工具可识别嵌套在大型重复区域中的CSV。基于T2T-CHM13参考基因组的分析显示,平均每个基因组含72个CSV,累计识别出1,247个CSV,涵盖128种结构特征,其中不少由串联重复介导,如DEL-INV-DEL、INVDUP-INV-DEL等。研究人员重点解析了与大脑发育相关的NOTCH2NL和NBPF基因的三种CSV单倍型,以及与脊髓性肌萎缩症相关的SMN1/2区域,成功组装了101种单倍型,明确了拷贝数和功能基因。淀粉酶基因簇区域也被完全解析,共识别39种单倍型,其中H1a.1、H3r.1等四种最常见,最长的单倍型含11个AMY1拷贝。
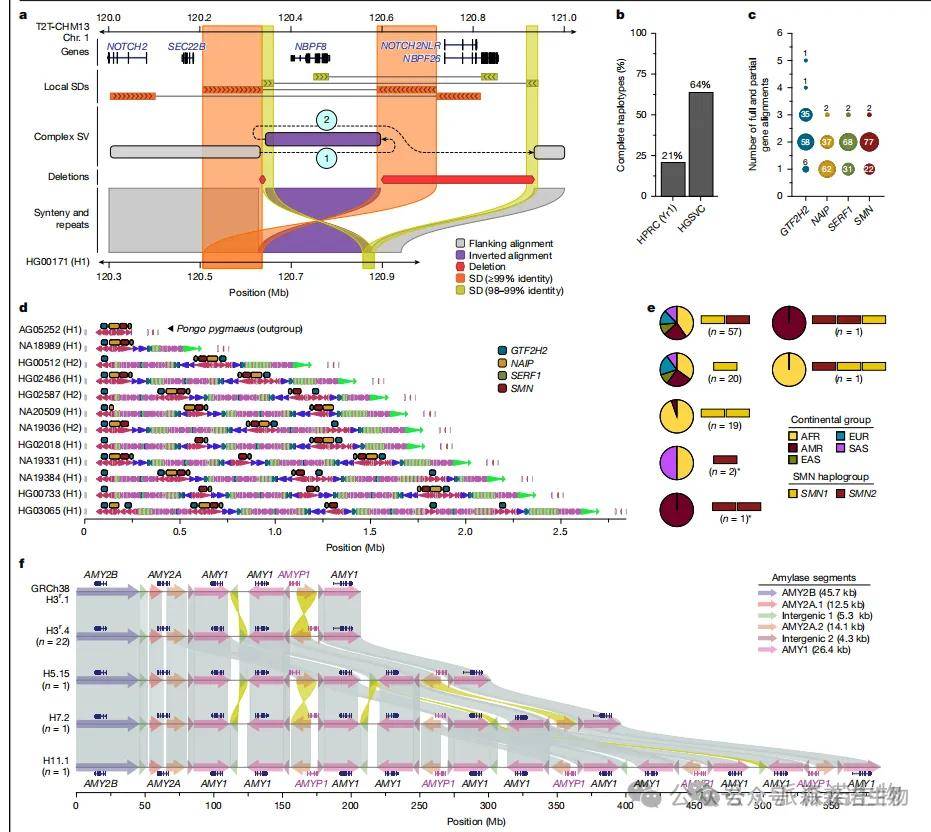
图5 人类基因组中复杂的结构变异
6.着丝粒研究
人类着丝粒是高度变异的基因组区域,由α-卫星DNA构成的高级重复单元(HORs)组成,长度和结构在不同个体间差异显著。研究人员利用Verkko和hifiasm两种算法,首次在65个基因组中高质量组装出1,246个完整着丝粒,平均每个基因组约19.5个。研究发现α-卫星阵列在长度上差异较大,并识别出4,153种新型HOR变体和阵列结构,部分染色体上出现阵列分裂现象。通过CpG甲基化分析,所有着丝粒均存在低甲基化区域(CDR),部分染色体呈现“双CDR”,提示可能存在双动粒结构。此外,约30%的α-卫星阵列中含有移动元件插入(MEI),主要为L1HS和Alu,大多分布在CDR之外。个别插入可能影响CDR结构,提示其在调控动粒定位和染色质结构中可能具有功能作用。
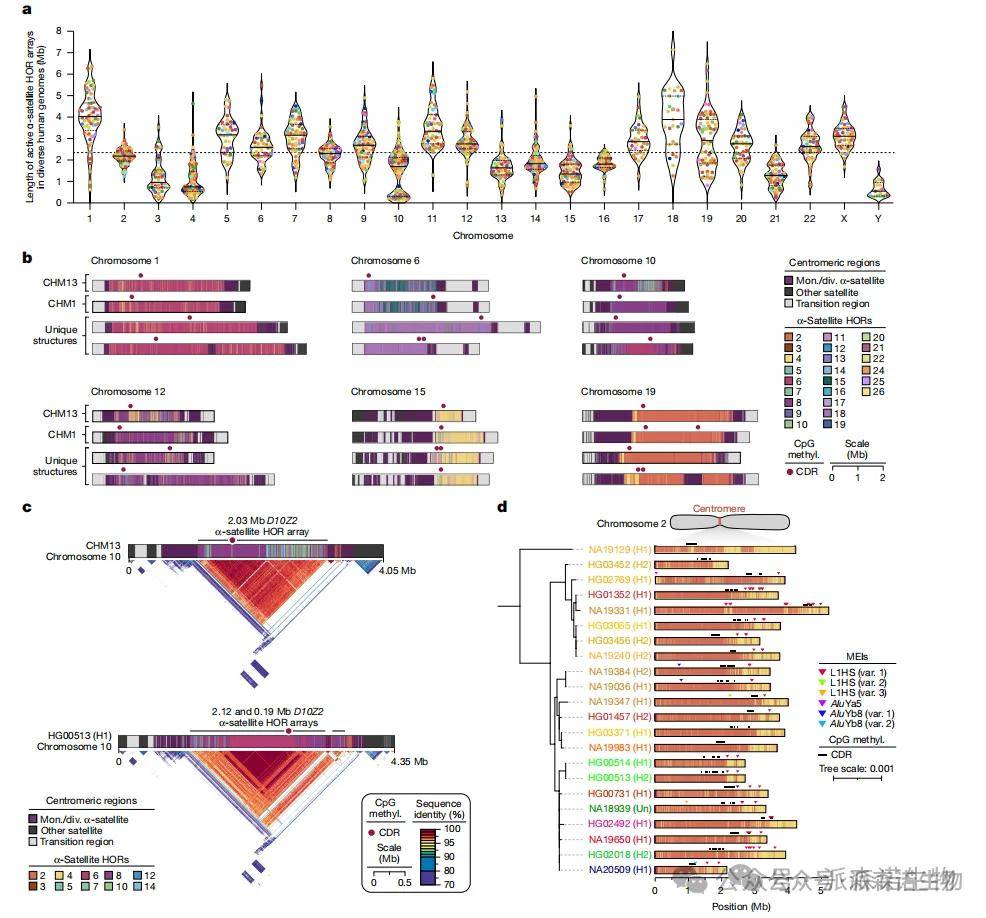
图6 人类着丝粒中的序列和结构情况
总 结
本研究通过对65个来自不同人群的人类基因组进行高连续性测序与组装,构建了130条近乎完整、无间隙的单倍型基因组,首次系统解析了复杂结构变异、着丝粒α卫星阵列的多样性、Y染色体结构、MHC区域的多态性,以及SMN1/2、AMY1/2等复杂基因座的结构特征。研究还结合泛基因组图谱,显著提升了短读长测序数据在复杂区域的基因型推断准确性,为疾病关联研究提供了更全面的遗传变异资源。


















