随着测序技术的进步,常规转录组测序凭借高通量、价格低、周期短等优势,成为入门级组学技术,广泛应用于农医学研究中。而发表高级转录组文章的过程中,对目标基因集的挖掘和讨论,往往才是重点。转录组数据量庞大,如何快速对目标基因集进行分析,从海量的分析结果中筛选关键目标基因,下面小派将对快速筛出候选基因提供以下通用性的建议和方法,您也可以结合其他筛选指标一起筛选出关键基因。
筛选思路
候选基因应该与老师所研究的生物学问题密切相关,并在某一生命过程中发挥核心的生物学功能、参与重要的通路。基因功能的背景信息可以来源于参考基因组注释、基因功能数据库(如GO、KEGG)注释或者相关研究的文献和综述等,获取可能相关的基因、GO和KEGG通路信息,然后在自己的测序结果中进行筛选。无论通过怎样的方式进行数据筛选,人为的解读和数据挖掘是必不可少的。
筛选方法
1、基于GO/KEGG富集分析结果
①打开GO富集结果*_download_enrichment.xls文件,在Term列中,用关键词逐一筛选与您研究相关的生物学功能,通常认为pvalue小于0.05的Term达到了显著富集程度,可以重点关注。获得相关显著富集的GO Term后可以在同表格里看到注释到相关GO Term的所有差异基因信息,示例如下:
![]()
②打开KEGG通路富集结果*_KEGG_download_enrichment.xls文件,在pathway列中,用关键词逐一筛选与您研究相关的通路,常认为pvalue小于0.05的通路达到了显著富集程度,可以重点关注。获取关注通路的富集情况后可以参照GO筛选方法,即可获得注释到关注通路的所有差异显著基因的信息。以查找“cGMP-PKG signaling pathway”为例,示例如下:
![]()
注:在某些实验处理条件下,差异表达基因可能较少,因而GO和KEGG通路富集结果较少,如果已经有关注的GO或KEGG通路,可直接从*.DESeq2.xls表中的GO列或KEGG列进行筛选,获取候选基因信息。
2、基于GSEA分析结果
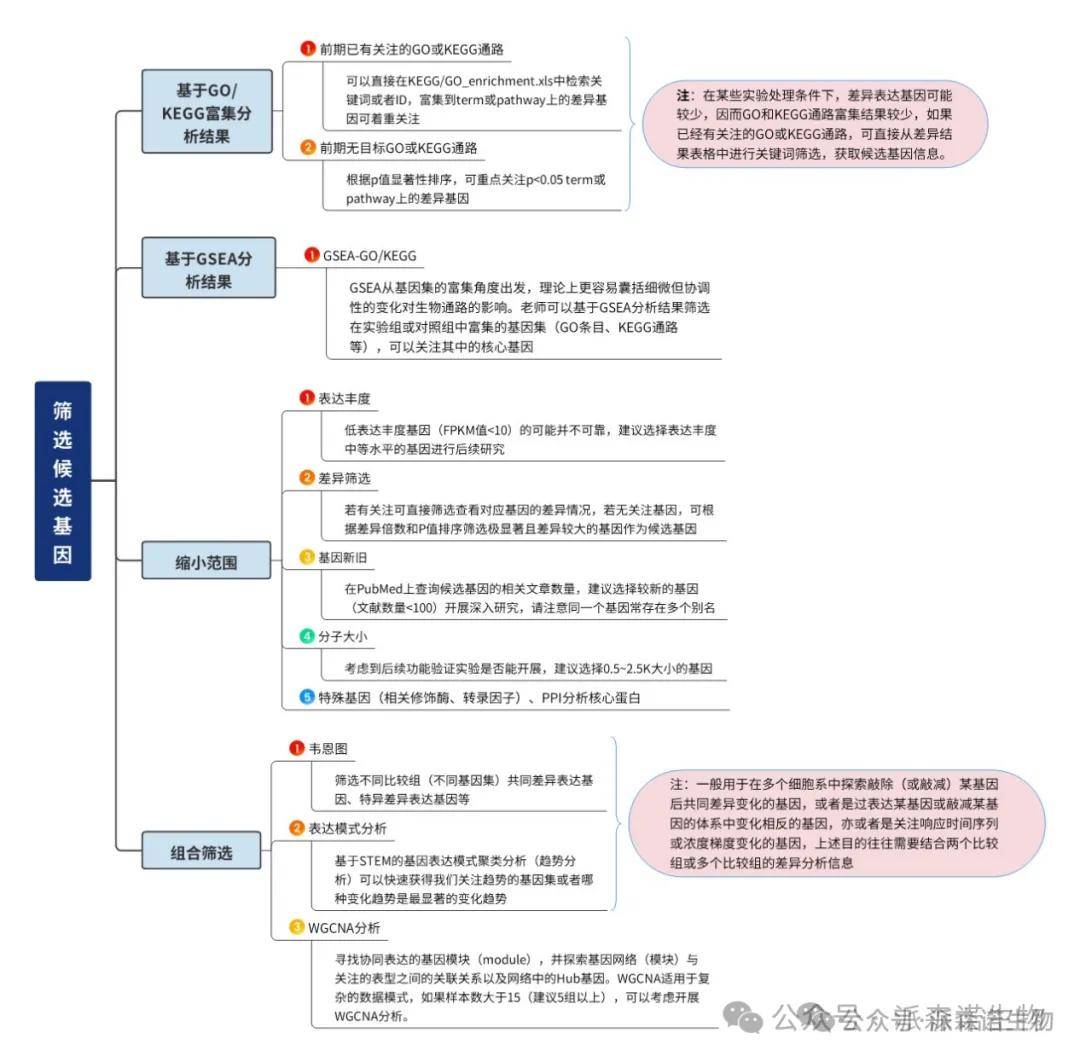
GSEA从基因集的富集角度出发,理论上更容易囊括细微但协调性的变化对生物通路的影响。我们可以基于GSEA分析结果筛选在实验组或对照组中富集的基因集(GO条目、KEGG通路等),然后可以关注CORE ENRICHMENT(对ES值有主要贡献的基因)。
3、缩小范围
表达丰度:受系统噪音影响,低表达丰度基因(FPKM值<10)可能并不可靠,建议选择表达丰度中等水平的基因进行后续研究。
差异筛选:若已有关注基因可以直接筛选查看对应基因的差异情况,若无关注基因,可根据差异倍数或者p值排序筛选极显著且差异较大的基因作为候选基因。
基因新旧:在PubMed上查询候选基因的相关文章数量,建议选择较新的基因(文献数量<100)开展深入研究,请注意同一个基因常存在多个别名。
分子大小:考虑到后续功能验证实验是否能开展,建议选择0.5~2.5K大小的基因。
特殊基因:可以通过转录因子家族分析或者蛋白质-蛋白质相互作用(PPI)网络分析筛选关键基因或者核心蛋白。
详情请点击:
Cytoscape软文:
【干货】如何使用cytoscape玩转网络图?(一)
【干货】如何使用cytoscape玩转网络图?(二)
【干货】如何使用cytoscape玩转网络图?(三)
4、组合筛选
有时候我们需要在多个细胞系中探索敲除(或敲减)某基因后共同差异变化的基因,或者是过表达某基因或敲减某基因的体系中变化相反的基因,或者是动物模型或细胞模型中用药后表达有回复的基因,亦或者是关注响应时间序列或浓度梯度变化的基因,上述目的往往需要结合两个比较组或多个比较组的差异分析信息,以下筛选方法供参考:
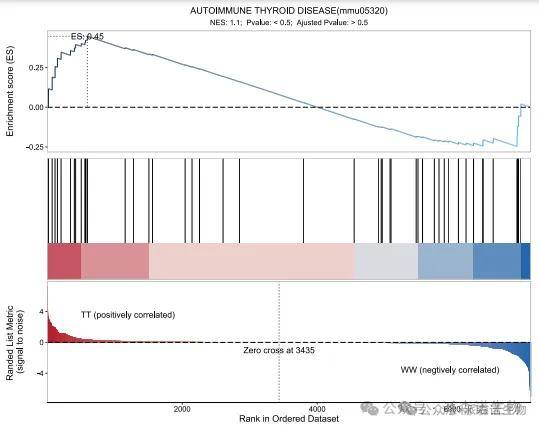
(1)韦恩图:
可以用韦恩图筛选不同比较组(不同基因集)共同的元素和特有元素,比如共同差异表达基因、特有差异表达基因等,并将这种筛选过程和结果可视化。灵活利用韦恩图可以通过一次筛选或多次筛选获取我们关注的核心基因集,然后可以对核心基因集进行进一步分析。比如利用韦恩图筛选某基因敲减组差异上调基因和过表达组差异下调基因的交集。
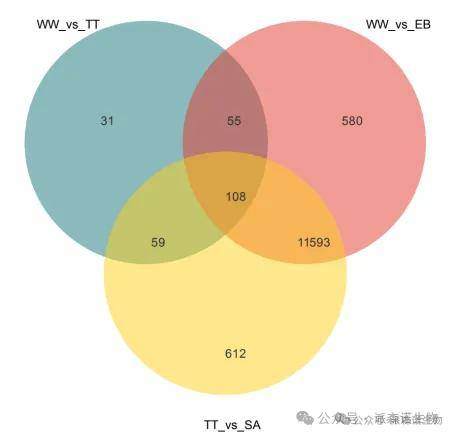
(2)表达模式分析:
当老师样本为不同时间节点、浓度梯度、治疗或用药前后的这种设置时,若进行两两比较筛选,当样本组较多时可能需要多次的相交筛选才能获得目标变化趋势的基因集。老师可以基于STEM(http://www.cs.cmu.edu/~jernst/stem/)的基因表达模式聚类分析或者常规趋势分析可以快速获得我们关注趋势的基因集或者哪种变化趋势是最显著的变化趋势。然后针对目标基因集进一步分析即可。
(3)加权共表达网络分析
加权基因共表达网络分析(WGCAN,Weighted correlation network analysis)是用来描述不同样品之间基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因集,并根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因或治疗靶点。WGCNA适用于复杂的数据模式,如果样本数大于15,可以考虑开展WGCNA分析。
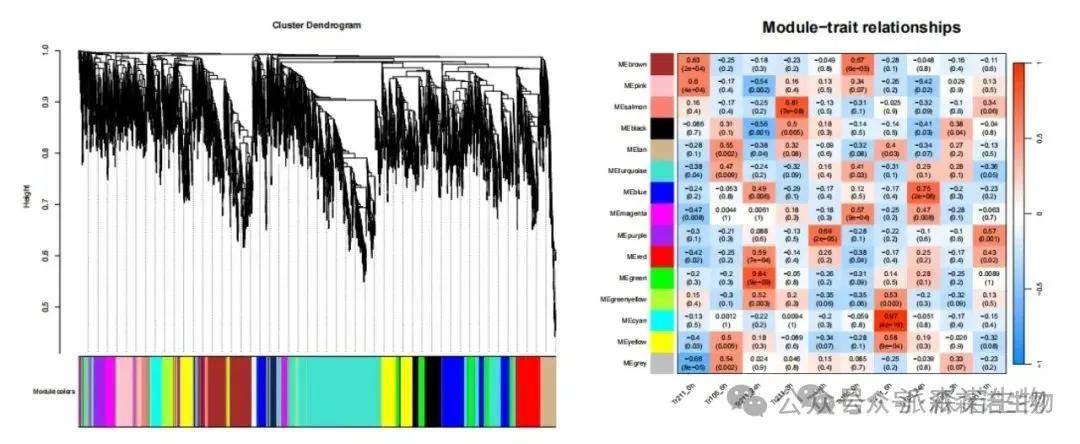
通过以上这几种分析方式可以找到目标功能、性状或不同实验处理中的核心基因。
那么如何基于转录组测序找到下游可验证基因,我们到这里就介绍完了,下方也绘制了我们今天讲到的分析思路路线图,有需要的老师可以收藏奥~当然,除了已经介绍过的方法外,将转录组数据与蛋白组/代谢组等组学数据进行联合分析也可以进一步缩小目标基因的筛选范围。
将多种分析方法及筛选路径结合使用,更能提升我们筛选到的核心基因的可靠程度,降低后续实验失败的概率。最后再重点强调一下,无论哪种方式挑选出来的基因,都需要尽量满足基于表达量和差异的目标基因筛选标准进行,老师们快来试试吧。