

2025-09-11

研究物种:小鼠
发表期刊:International Journal of Molecular Sciences
发表时间:2025年
影响因子:4.9
文章亮点
1. 研究策略新颖:传统智慧与现代技术的深度融合
“药食同源”概念的现代化阐释:文章没有孤立地研究单一草药,而是聚焦于一个具有深厚传统文化背景的类别——“药食同源”植物(如姜黄、生姜、大蒜、山楂、陈皮、丁香等)。这为开发低毒副作用、易于被接受的膳食补充剂或预防性药物提供了独特的思路。
多成分-多靶点-多通路”的整体视角:摒弃了传统单一靶点药物的研究模式,采用网络药理学这一前沿方法,完美契合了中医药整体治疗的理念。它系统地揭示了六种植物的协同作用机制,解释了为何复方或混合物比单一成分更有效。
2. 研究方法系统严谨:层层递进的多重验证
从海量数据中精准筛选:首先通过数据库挖掘,筛选出六种植物中的数百个活性成分,并预测出它们作用的数百个潜在靶点,构建了复杂的化合物-靶点网络。
核心靶点的巧妙锁定:通过将植物靶点与从疾病数据库(如TTD、DisGeNET)中获取的结直肠癌(CRC)相关靶点取交集,精准筛选出核心治疗靶点。并利用蛋白互作(PPI)网络、拓扑分析(Degree值)等生物信息学方法,进一步鉴定出如AKT1, TP53, VEGFA, CASP3, EGFR, MYC, JUN等最关键的靶点。
通路富集揭示作用全景:对核心靶点进行GO功能和KEGG通路富集分析,不仅验证了已知的癌症通路(如PI3K-Akt信号通路、癌症通路),还可能发现一些新的或次要的机制联系,全面勾勒出这些植物干预CRC的生物学过程全景图。
分子对接验证可靠性:文章通常不会止步于预测,会使用分子对接技术模拟核心活性成分(如槲皮素、山奈酚、β-谷甾醇等)与核心靶点蛋白(如AKT1, VEGFA)的结合构象和结合能,从计算结构生物学的角度验证预测结果的可靠性,极大增强了结论的说服力。
3. 研究结果具有重要的潜在应用价值
阐明协同作用机制:明确了这些植物通过多种成分协同作用,共同调节癌症相关的核心靶点和信号通路,从而抑制肿瘤细胞增殖、促进凋亡、抑制血管生成和转移等,为其传统功效提供了坚实的现代科学依据。
发现关键活性成分:识别出共有的关键化合物(如槲皮素、山奈酚),这些成分可能是发挥抗癌效用的物质基础,为后续提取、纯化以及质量标准化控制提供了重点方向。
提示潜在治疗靶点:筛选出的核心靶点不仅是植物起作用的关键,也可能成为未来开发新型CRC药物的候选靶点。
为预防和辅助治疗提供理论支持:由于研究对象是安全性较高的药食同源植物,该研究结论特别适用于结直肠癌的膳食预防、癌症高危人群的干预以及临床放化疗的辅助治疗策略开发,具有巨大的转化潜力。
研究背景
结直肠癌(CRC)是全球高发且高死亡率的恶性肿瘤,现有治疗方法如化疗副作用大,亟需寻找更安全有效的替代或辅助策略。 “药食同源”植物兼具营养与药用价值,安全性高,在预防和辅助治疗慢性病(如癌症)方面潜力巨大,但其抗CRC的科学机制尚不明确。传统单一靶点研究模式难以解释中药多成分、多靶点的整体作用特点。网络药理学与生物信息学方法能系统揭示“多成分-多靶点-多通路”的协同机制,完美契合该研究需求。
研究思路
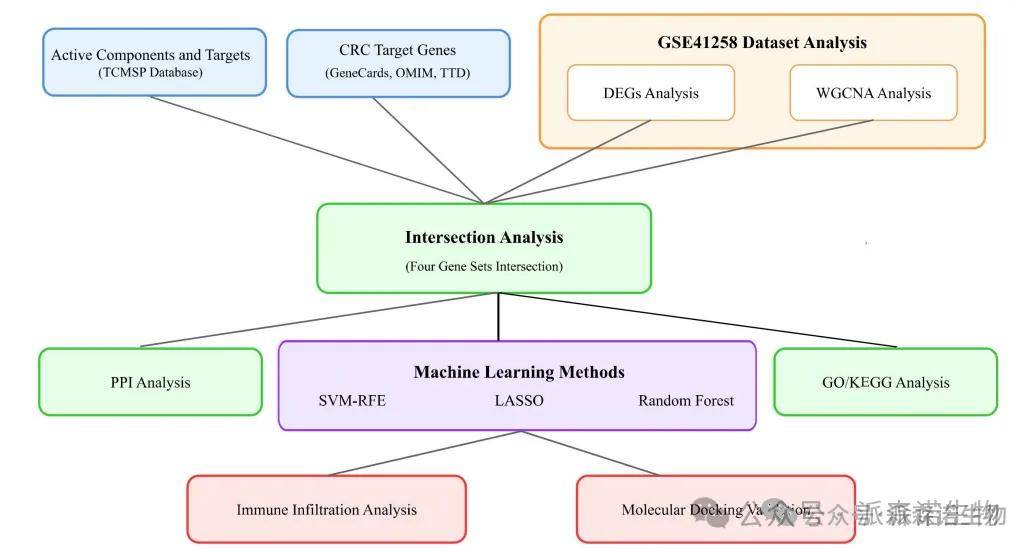
研究方法
网络药理学,机器学习
研究结果
1.筛选生物活性化合物和治疗靶标
使用TCMSP数据库,从六个药物同源植物中鉴定出303个活性成分和453个药物靶标。从Genecards,Omim和TTD等公共数据库中获得了总共11,593个CRC靶基因)。 GSE41258数据集的差异表达分析鉴定了1672个差异表达的基因,我们使用热图和火山图对其进行了可视化。使用GSE41258数据集,通过加权基因共表达网络分析(WGCNA)构建基因共表达网络,以识别与CRC相关的关键基因模块。相似模块的整合产生了12个独特的基因簇,通过树状图可视化。其中,棕色,蓝色和粉红色模块与CRC/NonCRC表型表现出牢固的相关性。统计评估表明,这三个模块中与CRC相关基因之间的基因意义(GS)和模块构件(MM)指标之间存在强烈的正相关性。将三个模块的基因合并以获得WGCNA结果。通过交点计算,确定了49个潜在的治疗靶标。这些结果表明,这六种草药可以通过多个靶标的多个活性组件进行协同作用。
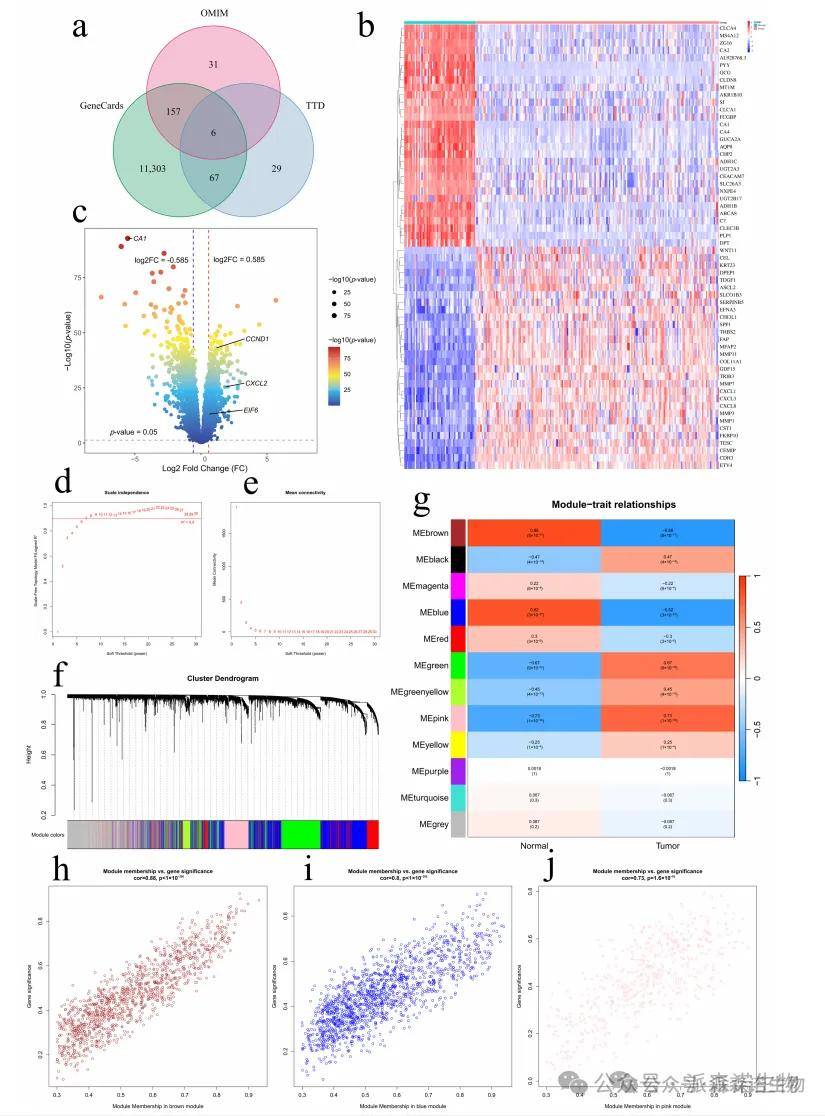
图1,识别与CRC相关的靶标。
2.建立蛋白质 - 蛋白质相互作用和药物 - 蛋白酶网络
使用STRING数据库构建了包含49个相交基因的PPI网络,并随后通过Cytoscape可视化(图3B)。此外,我们构建了一个融合了草药类型,活性成分和49个重叠靶基因的药物-直肠癌网络。
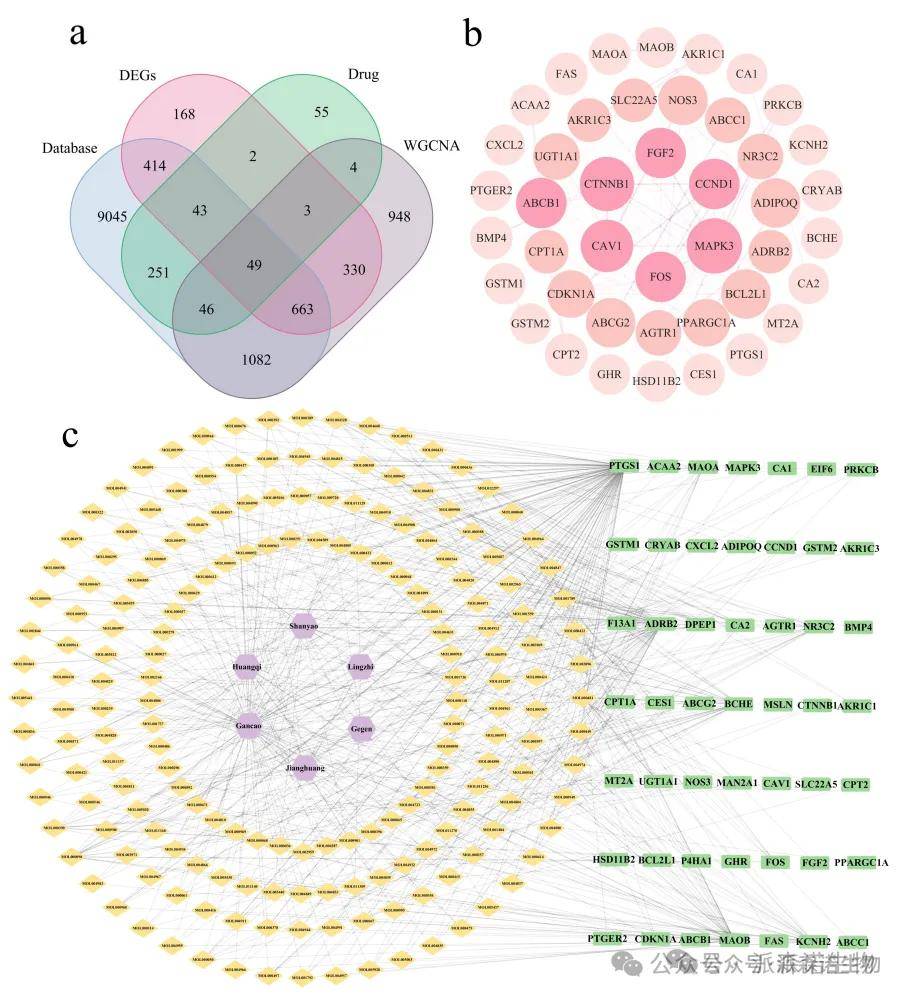
图2. CRC中治疗目标和相互作用网络的计算分析。
3.GO和KEGG功能分析
基因本体论(GO)富集分析表明,靶基因主要与生物过程(BP)相关,包括药物反应,脂肪酸代谢,酒精反应和激素反应。细胞成分(CC)类别包括细胞,小窝,膜筏,内质网腔,线粒体外膜和微绒毛的顶端部分。分子功能(MF)术语突出了类固醇结合,异种生物跨膜转运,单羧酸结合和各种脱氢酶活性。基因和基因组(KEGG)途径富集分析的京都百科全书显示,与癌症相关途径(包括大肠癌和其他致癌途径)的涉及与CRC病原体的既定机制一致。
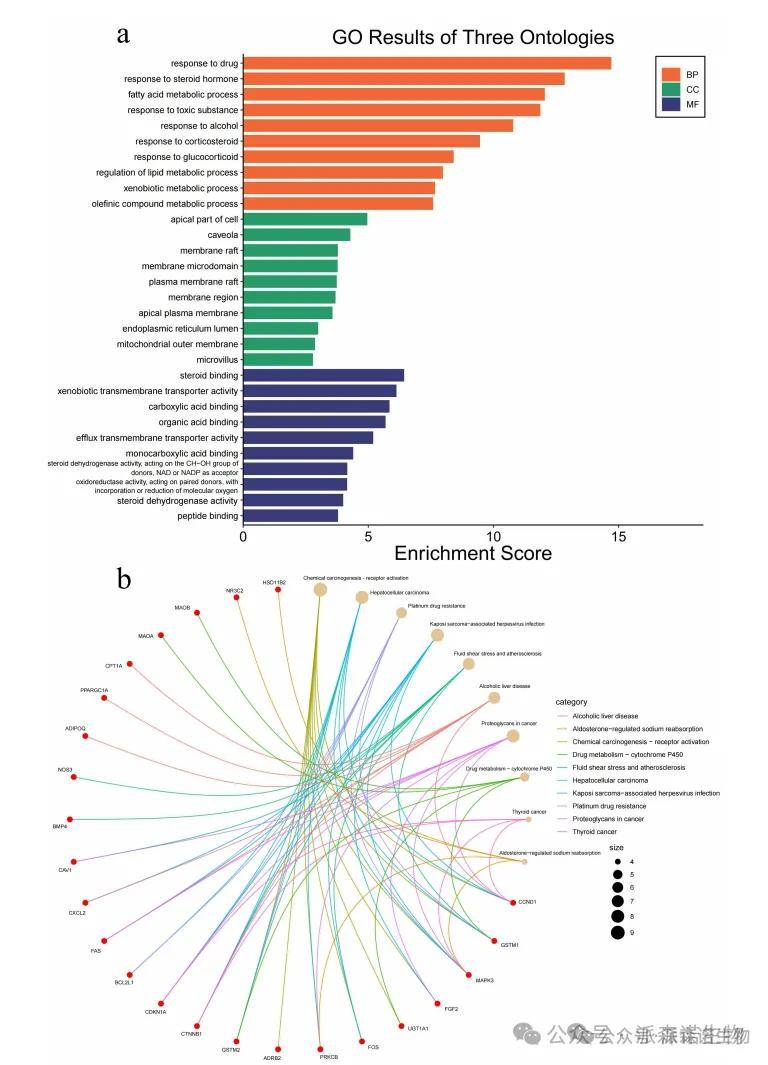
图3 对结直肠癌中六种传统中国草药的功能富集分析。
4.核心基因的识别和验证
使用三种机器学习方法,我们筛选了49个相交基因,以评估其将CRC样品与GSE41258数据集中的对照区分开的能力,最终识别基因。 SVM-RFE算法鉴定了十个基因,其中包括CA1,Ca2,NR3C2,CES1,CCND1,CCND1,CXCL2,BCL2L1,HSD11B2,EIF6和ADIPOQ。通过拉索分析,将9个基因(ABCC1,BCHE,BCL2L1,CA1,CCND1,CES1,CXCL2,EIF6和P4HA1)做为核心靶基因。
三个机器学习筛选结果的交叉分析最终确定了4个核心基因(CA1,CCND1,CXCL2和EIF6)。这四个核心基因在大肠癌中显示出显着的表达变化。CA1(碳酸酐酶1)被下调,表明肿瘤微环境中的pH调节改变了。 CCND1(G1/S特异性细胞周期蛋白D1),CXCL2(C-X-C基序趋化因子2)和EIF6(EIF6(真核翻译起始因子6)被上调,分别参与细胞周期调节,免疫反应,免疫反应和蛋白质合成。具体而言,CA1下调导致肿瘤微环境的酸化,从而为肿瘤进展带来了有利的条件。 CCND1上调促进G1/S相变,加速肿瘤细胞增殖。 CXCL2上调增强了炎症反应,重塑了肿瘤免疫微环境。 EIF6上调提高了蛋白质的合成效率,支持快速肿瘤细胞的生长。
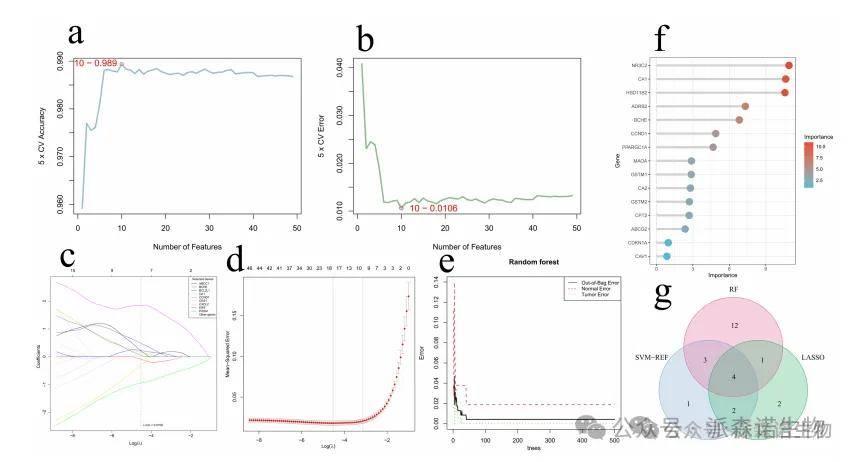
图4 通过机器学习方法识别Hub基因。
5.免疫细胞浸润和核心基因关联
CIBERSORT分析确定了结直肠癌和正常组织之间免疫细胞谱的显着变化。肿瘤组织显示自适应免疫细胞(包括浆细胞和记忆B细胞)的显着降低,伴随着活化的CD4+记忆T细胞的增加以及静息CD4+记忆T细胞的降低。在先天免疫细胞中,M0和M1巨噬细胞表现出较高的浸润水平,而肥大细胞表现出极化,其特征在于较少的静息细胞和活化细胞的增加。
相关分析揭示了核心基因与免疫细胞浸润之间的密切关联,尤其是与M0巨噬细胞,活化的CD4记忆T细胞和单核细胞之间的相关性。另外,核心基因对肥大细胞极化显示了调节作用。这些发现表明,核心基因可以通过调节免疫细胞动力学来重塑肿瘤免疫微环境。
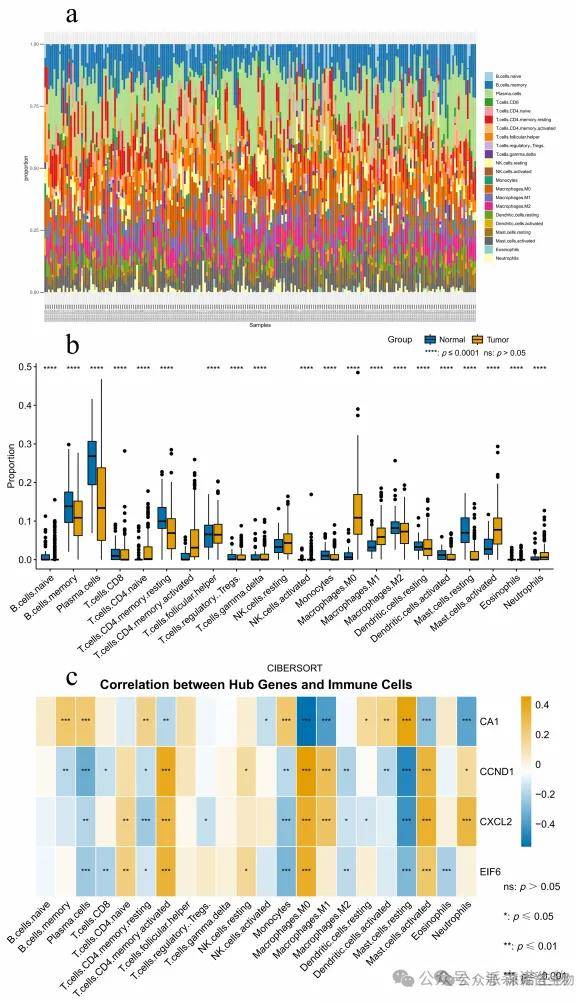
图5核心基因的免疫浸润分析。
6. 核心靶蛋白及其配体的对接分析
为了研究核心基因及其活性化合物之间的相互作用,我们进行了分子对接。低于-5 kcal/mol的结合能阈值用于确认结合亲和力。 CA1和CCND1结构对应于PDB文件1AZM和2W96,而CXCL2和EIF6结构是从Alphafold蛋白结构数据库中获得的。对接分析证实了所有核心靶蛋白及其配体之间的有利相互作用。我们说明了四种蛋白质的对接口袋以及提供了其相互作用的2D图。
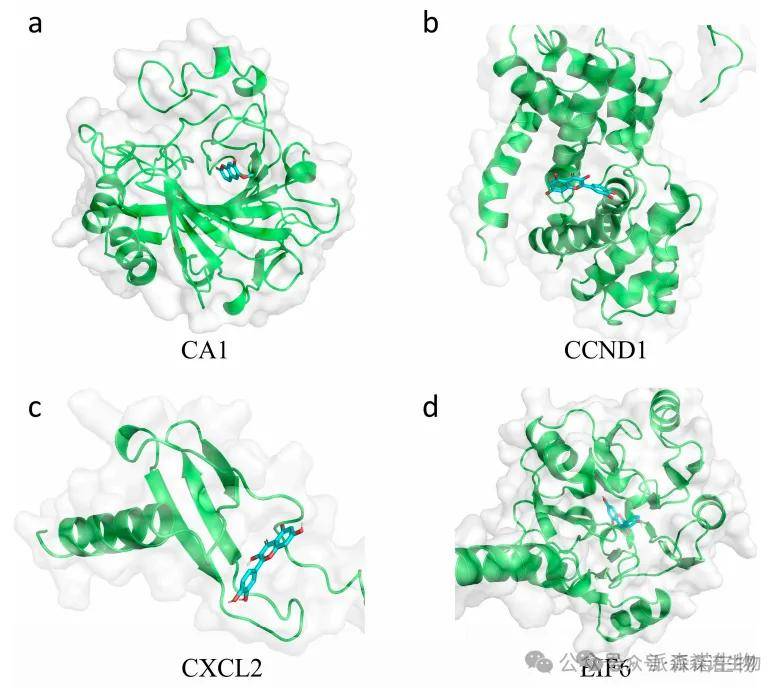
图6分子对接分析
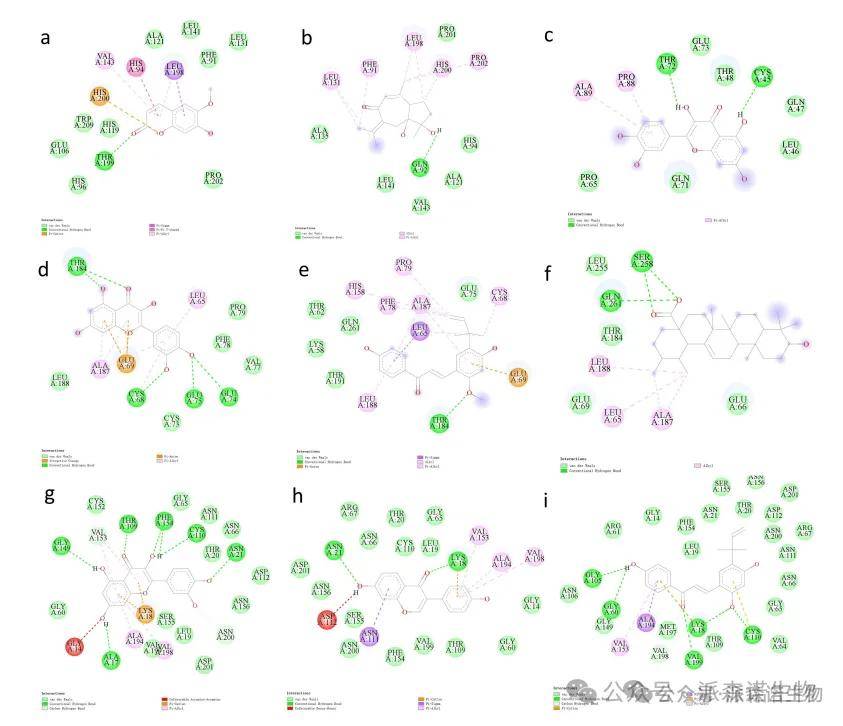
图7 目标核心蛋白及其相应配体分子的对接结果的相互作用图。
结 论
这项研究不仅从系统生物学的角度阐明了针对大肠癌的药物同源植物的分子机制,而且还为其合理的临床应用提供了理论上的支持。
参考文献
Zhao X, Xiu J, Yang H, Han W, Jin Y. Network Pharmacology and Bioinformatics Study of Six Medicinal Food Homologous Plants Against Colorectal Cancer. Int J Mol Sci. 2025 Jan 23;26(3):930. doi: 10.3390/ijms26030930. PMID: 39940699; PMCID: PMC11817456.


















