

2016-09-01
在上一期的“5分钟小课堂”中,小编介绍了宏基因组学大数据拼接组装的基本原理和操作工具,小伙伴们有木有亲自体验一把“拼图游戏”呢?同时,我们也已经提到,拼图获得的Contigs/Scaffolds序列将是后续一系列分析的数据来源。比如,各位小伙伴一定很想知道,这些序列各自具有怎样的生物学意义呢?也就是说,拼图揭示的这些微生物都在干什么?要想解答这个问题,就需要借助宏基因组学研究的“法宝”之二:功能注释(Annotation)!
1. 要想注释好,数据库寻宝
Contigs/Scaffolds序列经基因预测、ORF开放阅读框识别(Open Reading Frame)和蛋白翻译之后,就可以进行功能注释分析了。我们将基因/蛋白序列在特定的数据库中搜索比对,从而完成功能注释分析。常用的功能数据库主要包括KEGG、EggNOG和CAZy等。
1.1 KEGG数据库
KEGG数据库(Kyoto Encyclopedia of Genes and Genomes)[1]是最常用的功能注释数据库,其核心为生物代谢通路分析数据库(KEGG PATHWAY Database),以KEGG直系同源基因簇(即KO,KEGG orthologous groups)为基本单元,根据各个KO的具体功能逐层归类,并绘制代谢通路地图。
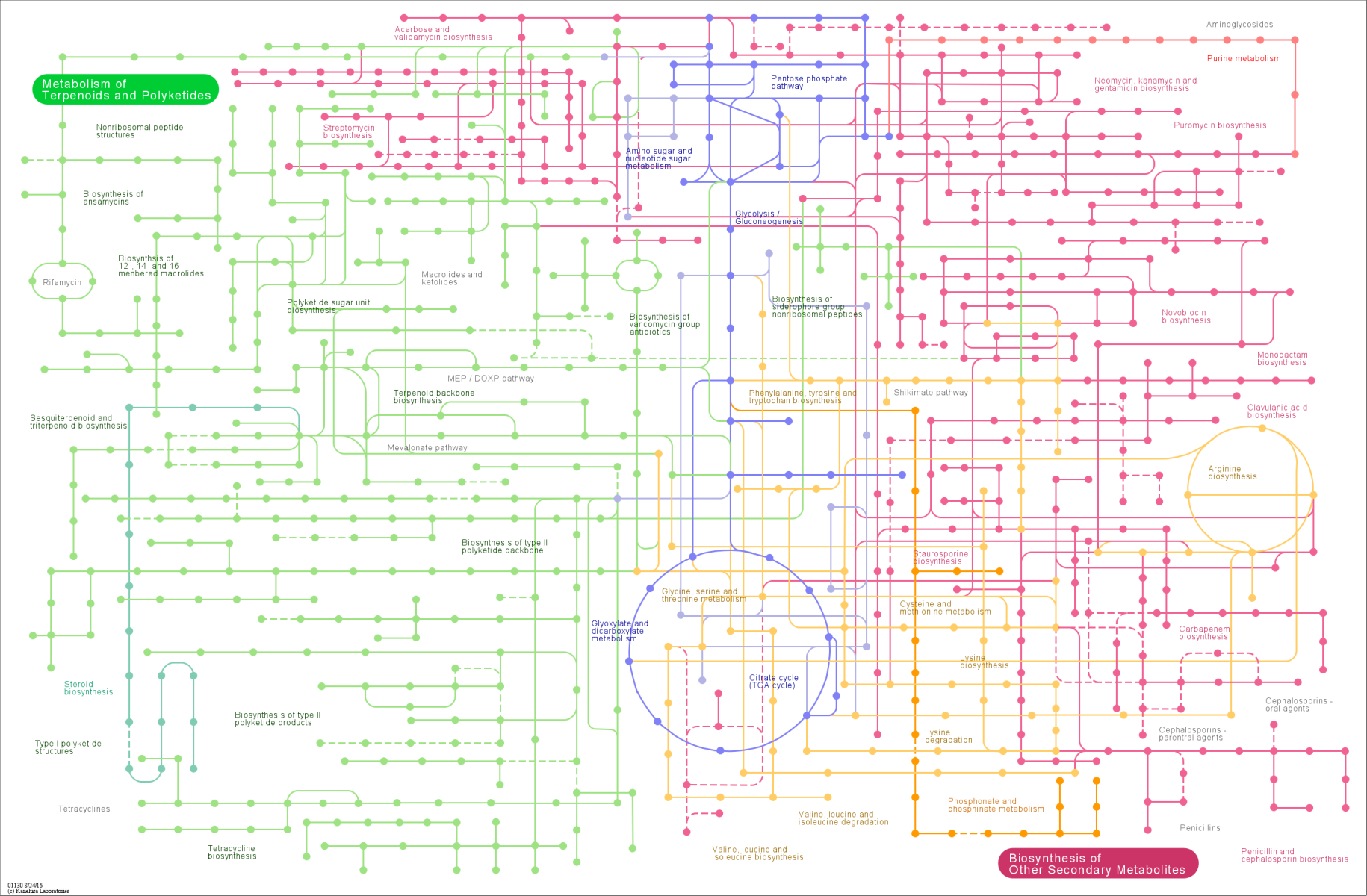
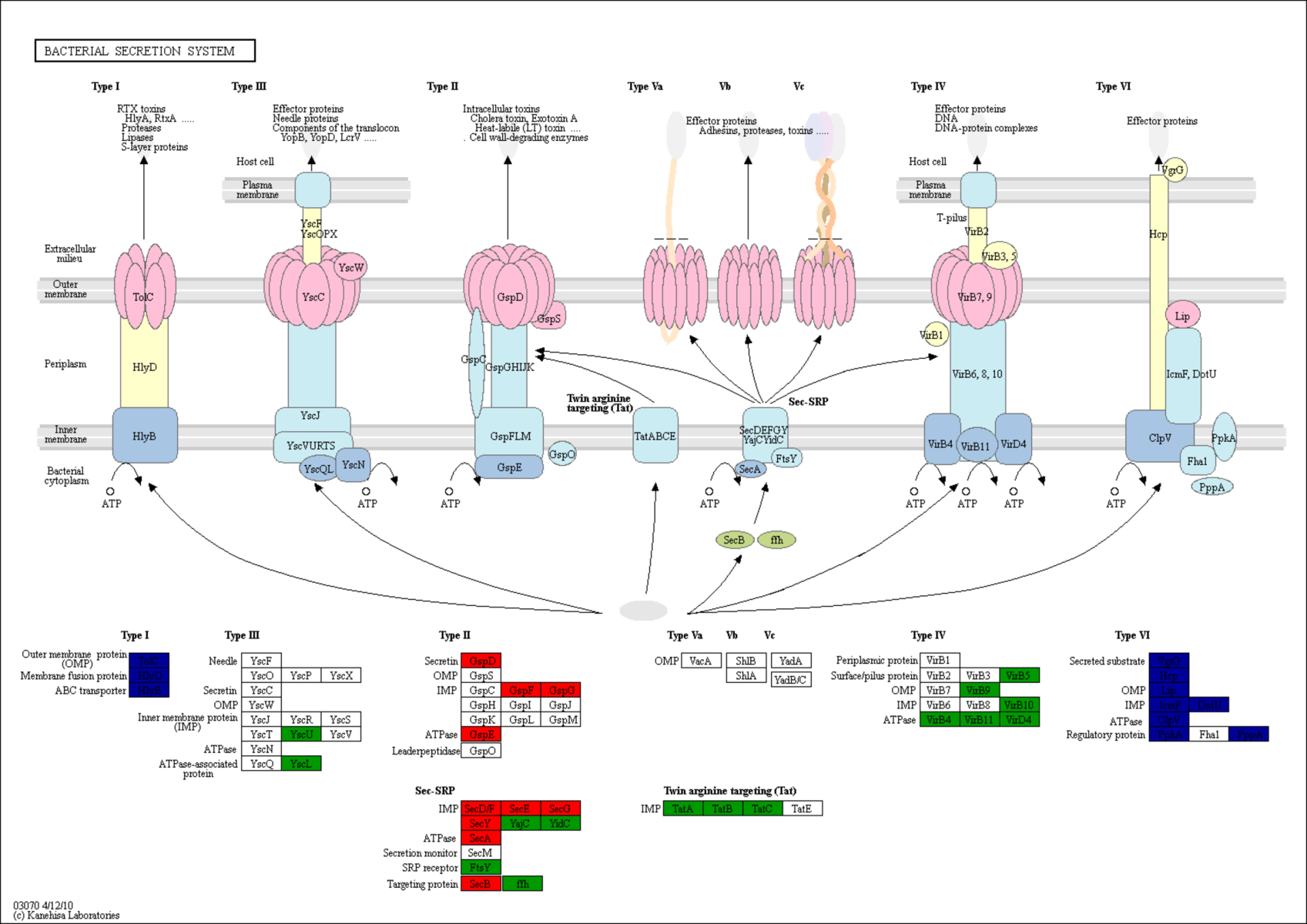
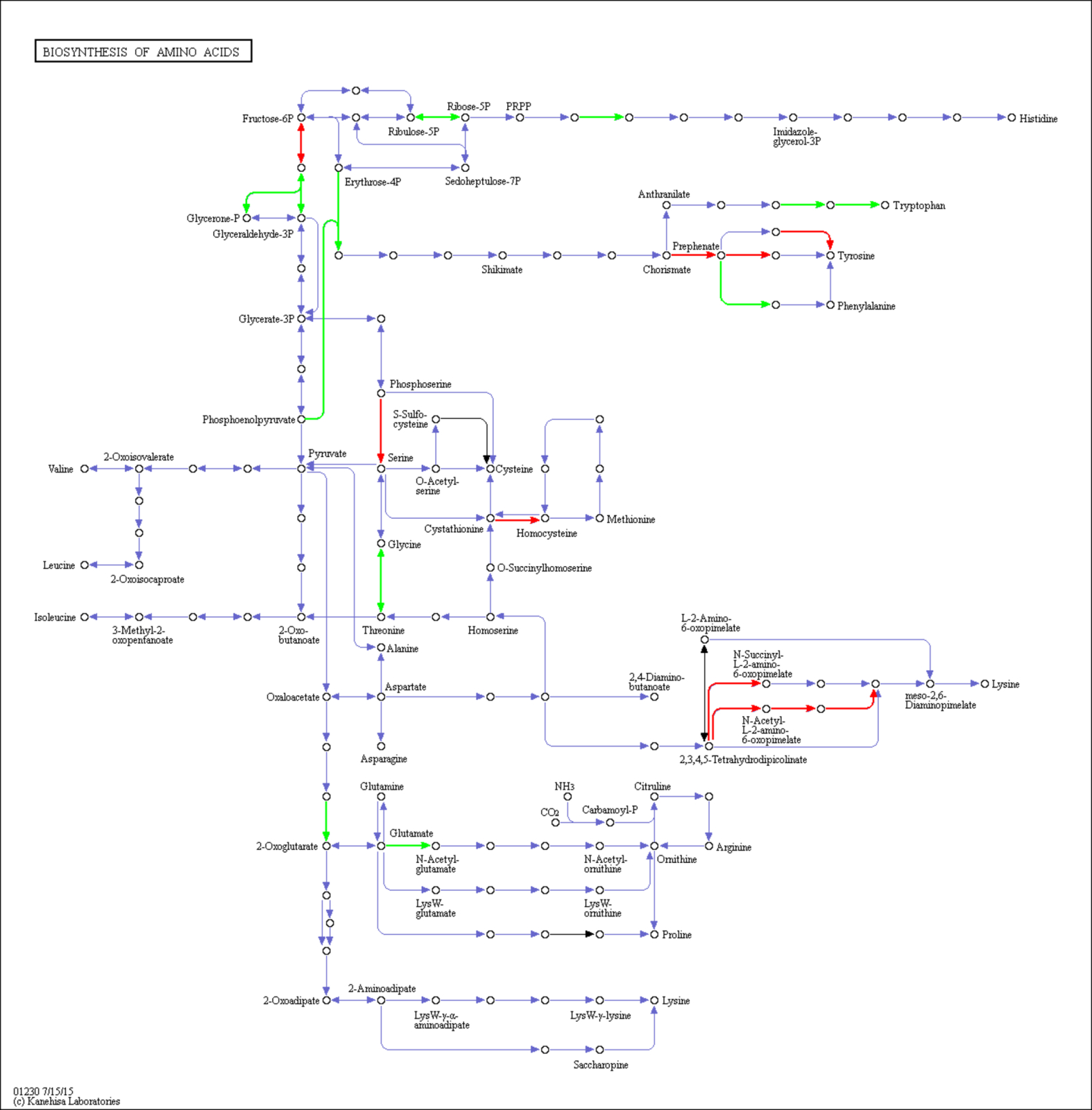
KEGG代谢通路地图示例
KEGG数据库的特色之一就是完善的代谢通路地图和注释说明。不仅如此,作为最常用的功能注释数据库,KEGG还提供了KAAS(KEGG Automatic Annotation Server)[2]和GhostKOALA(KEGG Orthology And Links Annotation)[3]等在线注释分析平台,我们只需要提交蛋白序列,即可获取相应的KO注释结果。

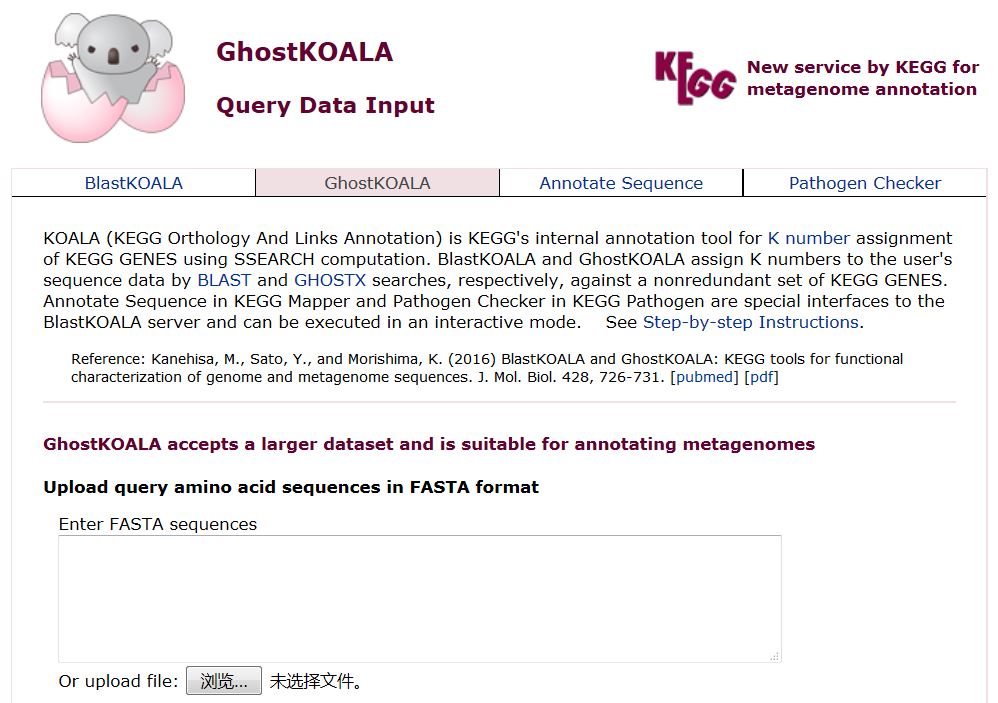
KAAS和GhostKOALA在线注释分析平台
1.2 EggNOG数据库
EggNOG数据库(Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups)[4]由欧洲分子生物学实验室(European Molecular Biology Laboratory,EMBL)所管理,目前已构建了接近200万个直系同源基因簇(Orthologous groups of genes)的功能注释信息。EggNOG数据库将基因功能分为25个大类,每一大类以一个英语大写字母代表。
EggNOG数据库主页截图
1.3 CAZy数据库
CAZy数据库(Carbohydrate-Active enZYmes Database)[5]成立于1998年,聚焦于降解、修饰或生成糖苷键(Glycosidic bond)的碳水化合物活性酶,是研究相关酶类的专业数据库。
CAZy数据库主页截图
除了上述常用数据库外,还有PHI病原与宿主互作数据库、VFDB病原菌毒力因子数据库、MvirDB生物防御数据库、CARD抗生素抗性基因综合数据库等等各种功能注释数据库,它们可以帮助我们充分挖掘宏基因组学数据,全面阐释菌群功能特性。
2. 功能注释丰度谱分析
在获得菌群的功能注释信息后,我们就可以对各样本的功能代谢谱进行一系列更深入分析啦!以KEGG数据库注释结果为例,我们可以统计菌群中各类代谢功能的数量:
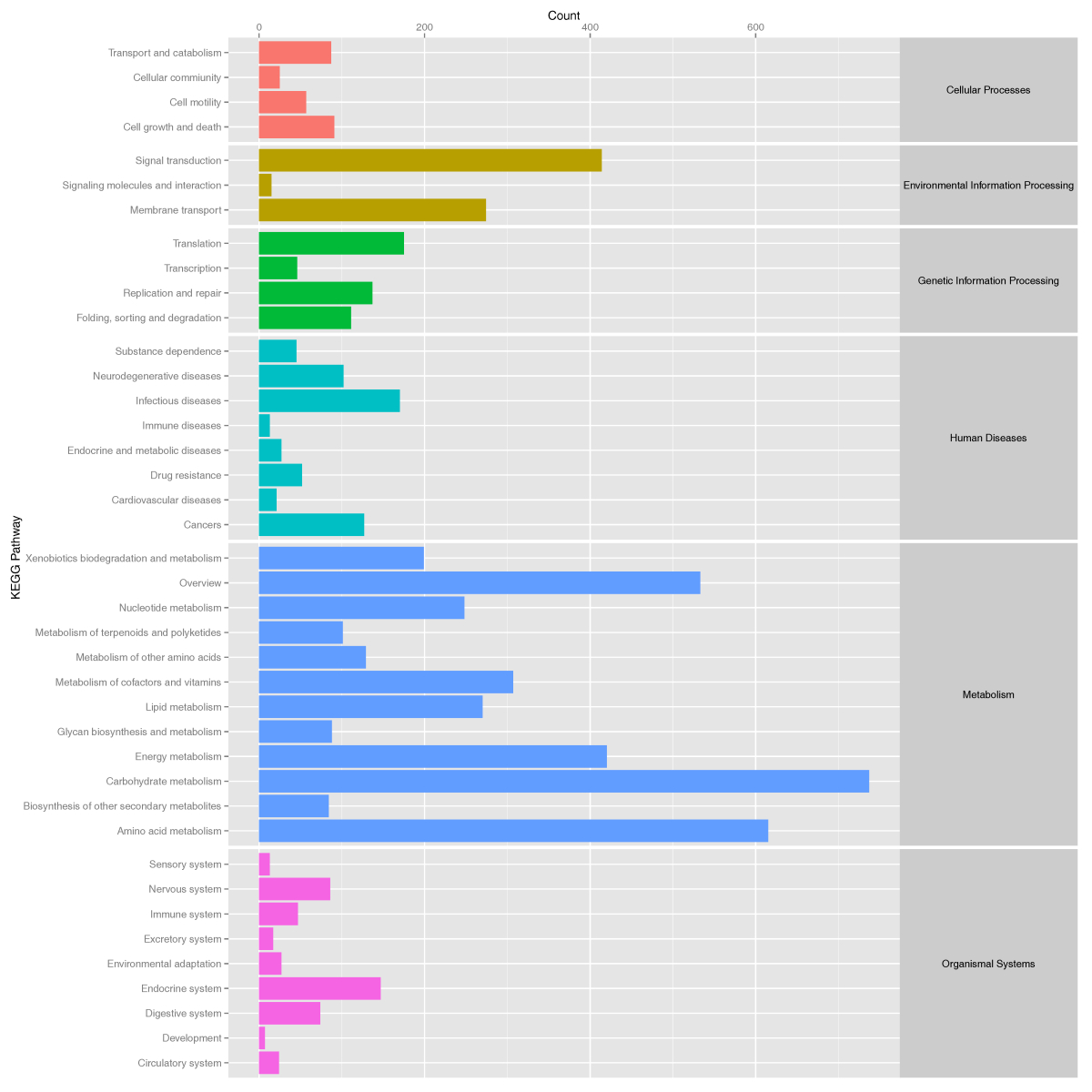
KEGG第二等级代谢通路的注释结果统计图
我们还可以进一步分析两样本(组)间共有和独有的代谢通路,或是它们各自富集的代谢通路:
共有/独有代谢通路图
代谢通路富集分析图
我们还可以根据代谢通路富集分析进行聚类分析,并绘制热图:
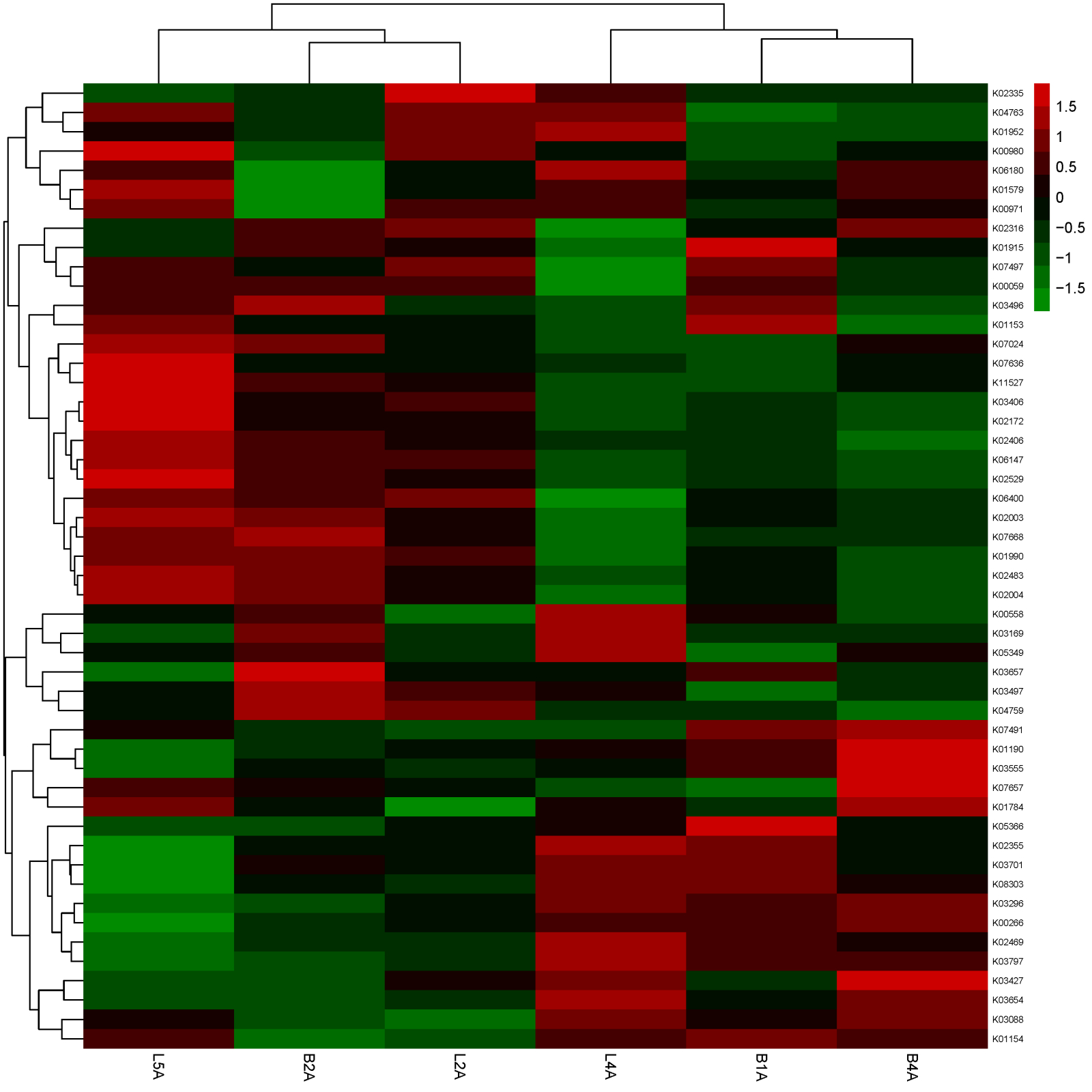
KO聚类分析热图
是不是感觉分析结果立马高大上了?小编告诉您,上述这些还只是基础分析,只要好好利用功能代谢谱的数据,有可能发现更深入、更有科学意义的研究结果!
3. 物种组成谱的注释分析
除了对宏基因组的代谢功能谱进行注释分析,我们还可以对物种组成谱进行分析,通过将Contigs/Scaffolds序列与NCBI-NT数据库进行BLASTN比对,可以获得宏基因组的精细组成信息。与基于rRNA基因部分可变区/全长的菌群组成和多样性普查相比,宏基因组学物种组成谱分析往往可以根据特定微生物物种所独有的标记基因,在种以及种以下更精细水平(如菌株水平),以“高分辨率”展现菌群的组成结构:
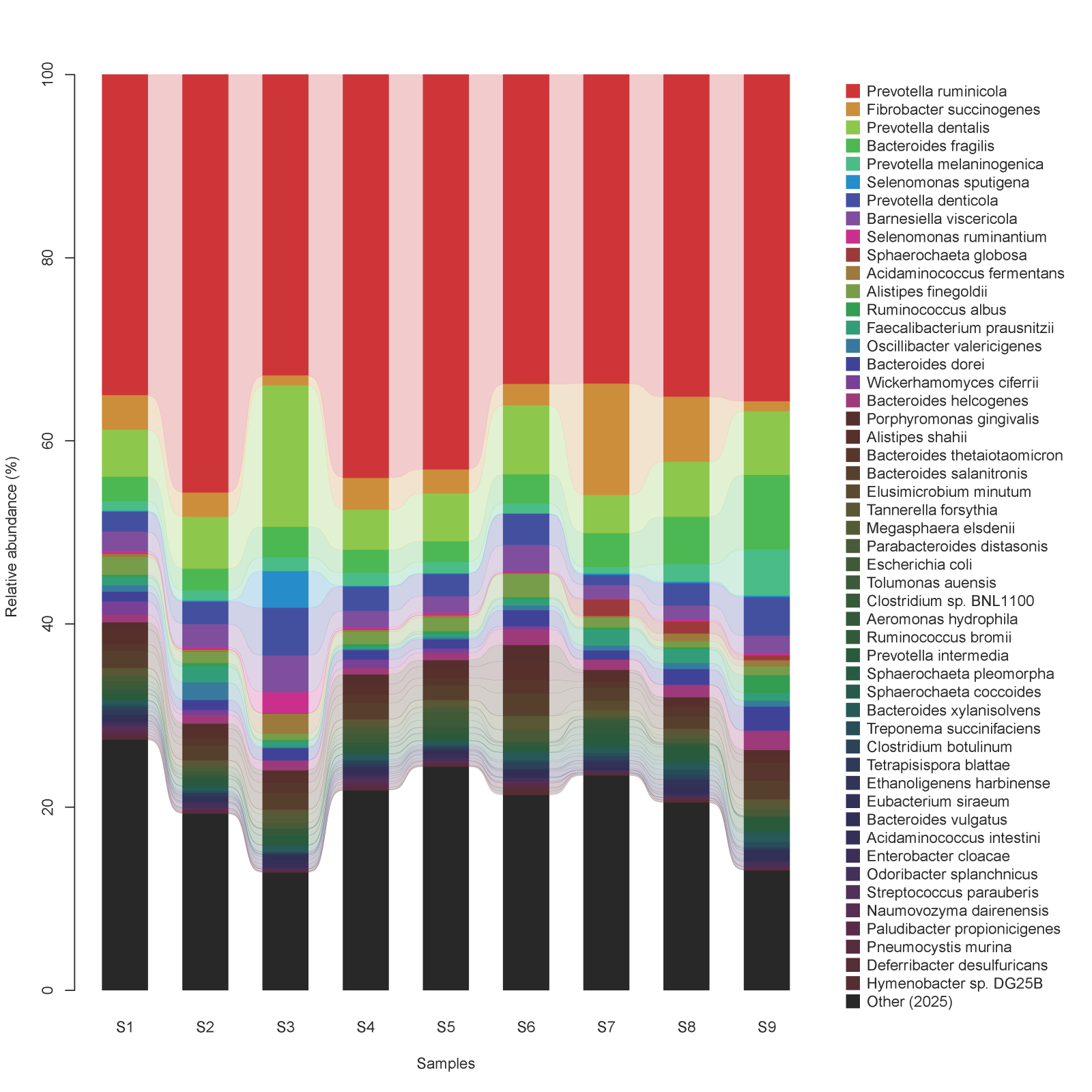
种水平的宏基因组物种组成谱分析图
我们还可以进一步评估宏基因组的功能丰度谱和物种组成谱是否具有一致的共性,并且通过关联分析,量化两者之间一致性的高低:
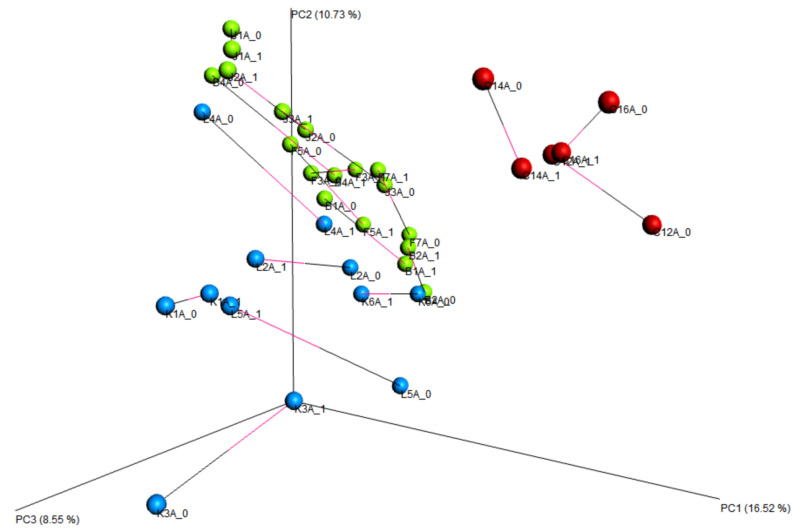
功能——物种一致性分析图
4. 结语
这一期的小课堂中,我们主要介绍了宏基因组功能和物种注释的方法,和相关的一系列数据分析结果。通过这些分析,我们不仅可以在种以及种以下的精细水平揭示“谁在宏基因组里?”,更能够阐明“它们在这里做什么?”。通过解答这两个层面的问题,我们可以对宏基因组的结构和功能有更全面的认识。
在获得宏基因组的功能丰度谱和物种组成谱后,如何进行更深入的数据挖掘并筛选生物标记物呢?欲知后事如何,且待下回分解,敬请各位小伙伴保持关注哦!
附:【5分钟小课堂】后续预告
l 茫茫菌群,谁是天使,谁是元凶,谁又是围观路人甲?
l 菌株水平的超高分辨率解析,宏基因组学就是这么高大上!
参考文献
1. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32: D277-D280.
2. Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35: W182-W185.
3. Kanehisa M, Sato Y, Morishima K (2016) BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J Mol Biol 428: 726-731.
4. Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, et al. (2008) eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res 36: D250-D254.
5. Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42: D490-495.


















