

2015-10-30
人体肠道微生物在宿主代谢、免疫和药物反应中扮演重要角色,与人类健康息息相关,然而每个菌种包含很多不同菌株,这些菌株编码的基因和每个基因的拷贝数可能差别很大,这种种内变异赋予不同菌株潜在的功能差异,因此不能单从菌种组成推断菌群功能,物种水平的比较分析可能无法获得样品间重要的功能差异。
Sharon Greenblum等人以之前研究中的109个健康、肥胖、炎症性肠病(IBD)个体的肠道微生物宏基因组样本为实验对象,运用新建立的分析方法直接从109个样品的宏基因组数据估算给定样本中的微生物菌种的每个基因的拷贝数,进而发现样本间的拷贝数变异。研究者总共将超过24.5 亿条的 75 bp 短序列map到 235个参考基因组(分为96 个genome clusters)上,运用建立的分析方法估计了几千个KOs的拷贝数,并利用获得的拷贝数估计的数据集,探索了肠道这一高度复杂的生态系统中的中性和适应性的变异,将宏基因组水平基因丰度的不同与基因组水平的变异联系起来。
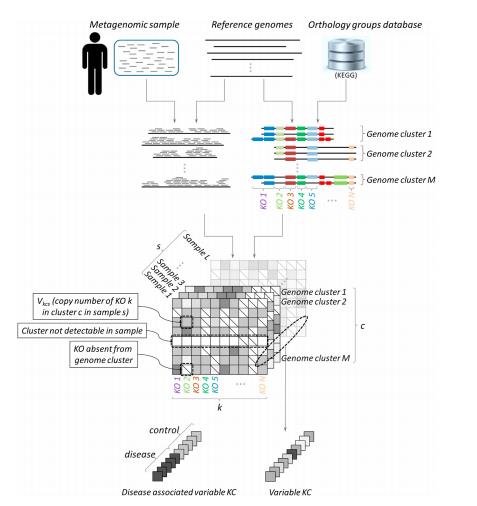
分析方法示意图
研究发现,拷贝数变异在肠道环境十分普遍,有的物种超过20%的基因都有显著的拷贝数变异,这些变异或许可以解释之前很多研究中出现的物种水平变化趋势和基因水平变化趋势存在差异的现象。通过分析研究者共检测到38个genome cluster的735个highly variable KCs,并检测到40个 genome clusters的 5,004个set-specific variable KCs。研究者发现每个cluster的highly variable KCs数变化很大,例如在Roseburia intestinalis cluster中多达47个(占此cluster全部KCs的4.05%),而平均每个cluster的highly variable KCs占1.79%。此外,很多反映适应性的变异为set-specific variable KCs,变异不显著且仅在少量样本中出现。
研究者将每个cluster的variable KCs与cluster中已测序的参考基因组的已知变异进行比较发现,数据库中的参考基因组并未包含全部的种内变异,同样,研究所用的样本可能也不包含很多参考基因组中的变异。
本研究发现运输功能相关的KCs非常倾向于高拷贝数变异,即成为highly variable KCs,相当大比例的肠道微生物物种中运输功能的基因拷贝数存在灵活性,特定的运输功能的基因可能会促进微生物对肠道环境的适应性。此外,大多数set-specific variable KCs似乎与物种对环境的反应和与环境的相互作用有关,显示了肠道菌群强大的适应性潜能。研究结果显示,尽管变异是可能是个体性的,但某些(如与环境适应性相关的)基因和功能可能普遍倾向于发生变异。
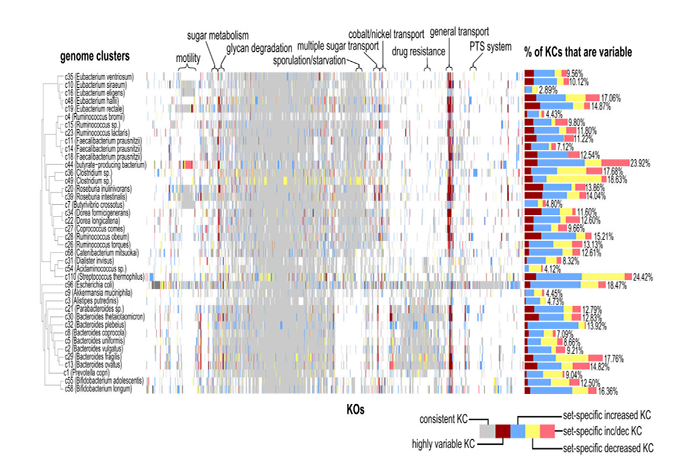
Variable KCs示意图
一些变异可能表现了对特定宿主表型的适应。研究者对在健康个体和肥胖或IBD个体间显著不同的可变KCs进行研究,最终发现24个KCs的拷贝数与IBD显著相关,3个KCs的拷贝数与肥胖显著相关(FDR<0.05)。该发现进一步揭示了单物种对疾病的作用,例如, Roseburia inulinivorans中编码主要药物外排蛋白的基因(K08217)的拷贝数的增加与IBD久治不愈高度相关。
研究者使用回归分析将每个cluster的拷贝数去卷积成为数据库中所含菌株的线性组合。在有很多已测序基因组的cluster中,大多数样本的拷贝数可以用已知菌株的线性组合表示;而在只有少数菌株基因组序列已知的cluster中,有时只有部分拷贝数变异可以被表示。为进一步比较样本间拷贝数变异情况,研究者使用了主成分分析,此分析揭示了每个cluster内的复杂的群落结构,各样本间差异明显,表明个体化变异普遍存在。
本研究证明了种水平的研究导致的功能信息缺失的程度,有利于促进菌株水平微生物群落结构的研究,表明了分析微生物群落的种内变异对理解物种组成与群落功能之间的复杂关系非常重要。此外,本研究采用建立的分析方法定量表征了肠道微生物的变异,获得的数据集和研究方法可作为未来人类微生物或其他环境中微生物组成变化研究的宝贵资源。
本文由市场部技术支持 王红整理


















