

2017-09-20
可变剪切事件的发生增加了后生动物的转录组和蛋白组的多样性。但是,对于可变剪切在单细胞中的活跃程度和功能,目前还知之甚少。美国遗传医学研究所的主任Jef D. Boeke教授带领团队利用单分子全长测序技术,通过一个开放性软件SpliceHunter来发掘裂殖酵母减数分裂时期的转录组特征,发表在《Genome Research》,影响因子11.922。测序发现在裂殖酵母的减数分裂时期,发现17,669个异构体,其中发生了14,353个可变剪切事件。另外检测到了770个新转录本和53个预测蛋白,表明与其他物种的同源性和理论的稳定结构遗传。并且报告了异构体中可变剪切的复杂性,包含683个分子间区共同联系的内含子对。对这些因素的评估表明主要的新异构体并不都是特殊条件下形成的可译转录本,但是这些评估结果和生物学功能性新转录本是一致的。研究结果突出了裂殖酵母的性发育过程中异构体水平的多样性和动态变化。
实验发现4h之后,裂殖酵母的减数分裂开始,在6h的时候完成减数分裂。实验选取0-10h的时间段,每两个小时取样一次,每个时间点两次重复。对AS事件类型定义为8种:外显子跳跃,包含外显子,内含子保留,外显子包含内含子,可变受体,可变供体,可变受体/供体,新外显子。示例图见图1。
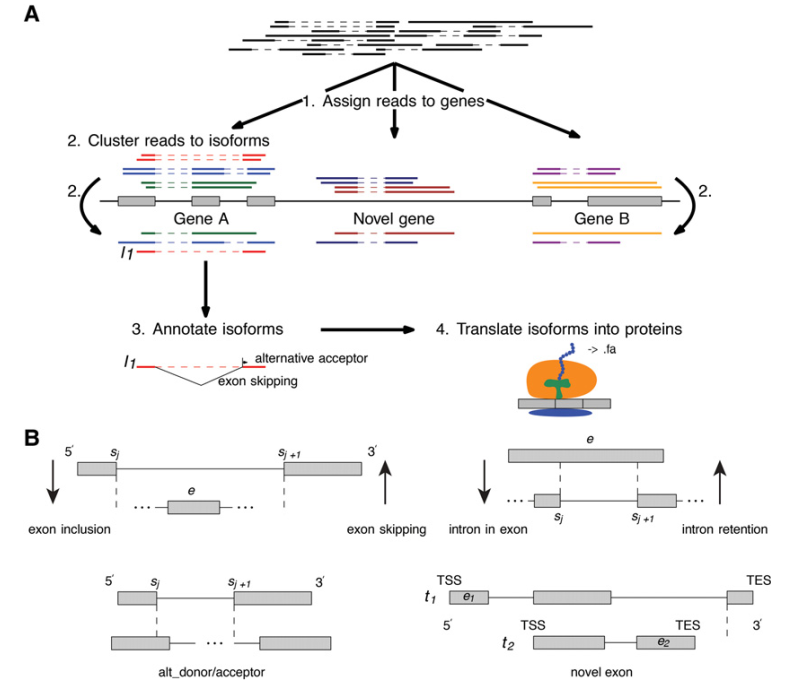
图1 PacBio测序的转录组分析总结
三代测序共获得2,266,791个CCSreads,其中包含1,322,840个FL CCSreads。过滤后鉴定出424,511个Iso-seq reads (Read of insert)(图2A)。CCSreads的平均长度是1285bp,比之前的文章结果要略长。FL CCS reads和Iso-seq reads的平均长度是1,094bp和1,178bp,长度分布不随时间点发生变化(图2B)。PacBio测序发现了裂殖酵母的6,199个基因(所有基因的90%左右)。每个基因的FL CCS reads的平均覆盖次数是71(图2C),许多基因覆盖次数超过100,有利于对新转录本进行深入研究。利用PomBase定义(非)编码RNA和假基因,发现了4,993(97.1%)个编码蛋白基因,1,121(73%)个非编码RNA基因和18(62.1%)个假基因。对于FL CCSreads来说,97.4%比对到了编码蛋白基因,这代表了所有获得基因的80%,表明编码蛋白基因的高覆盖率(图2D)。有趣的是,发现编码蛋白的基因和ncRNA趋势相反,RNA分子比例在减数分裂中期先减少,在后期又增加(图E)。尽管大部分的reads含有0或者1个内含子,但是还有190,101个reads有多重内含子(图2F)。另外统计了reads的5’或者3’端与对应的基因转录本起始位置(TSS)或者转录本终止位置的距离(TES)。从平均水平看,Reads在注释基因的5‘端的上游延伸了232bp(图2G),3’端的下游延伸了188bp(图2H),暗示可变的5’和3’UTRs。
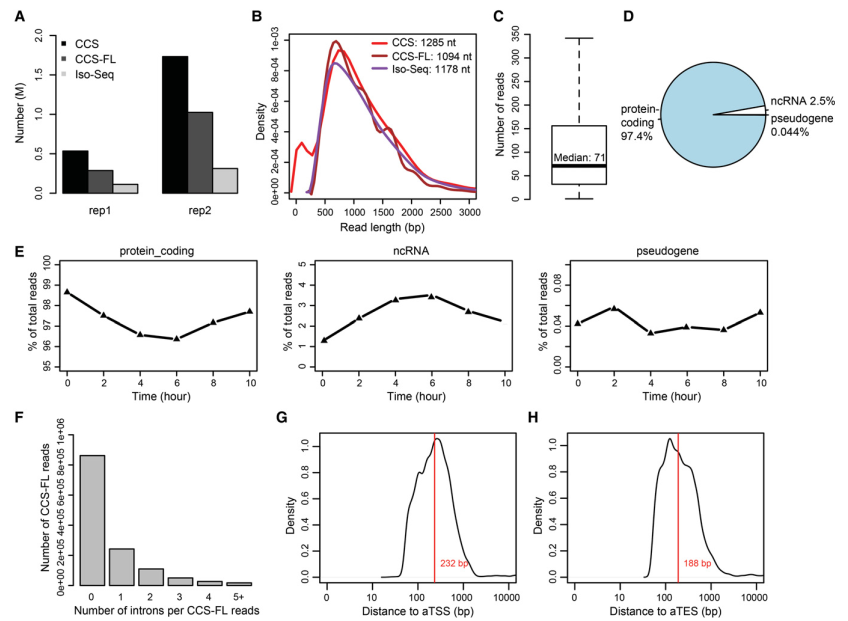
图2 PacBio reads的一般特性
研究发现SPAC12B10.05(icp55)有59个明显的聚腺苷酸mRNA异构体,包含了不同的AS剪切事件类型(图3A)。在S.pombe中发现的AS类型的实例如下:可变剪切受体类型,SPCC1281.08(wtf11);包含外显子类型,SPBC1703.10(ypt1);多重外显子跳跃类型,SPCC1235.11(mpc1);外显子包含内含子类型,SPAC144.02(iec1);内含子保留类型,SPAPB8E5.05(mfm1)和新外显子类型,SPAC1296.03c(sxa2)(图3B)。还发现,一些新转录本对应的reads数不比注释转录本少,甚至更多(图3C)。很多新异构体的剪切模式和相应的注释异构体有密切关系,一些新的异构体和相应的注释异构体相比,展现出明显的时间进程模式。表明新异构体可能被暂时性差异调控。
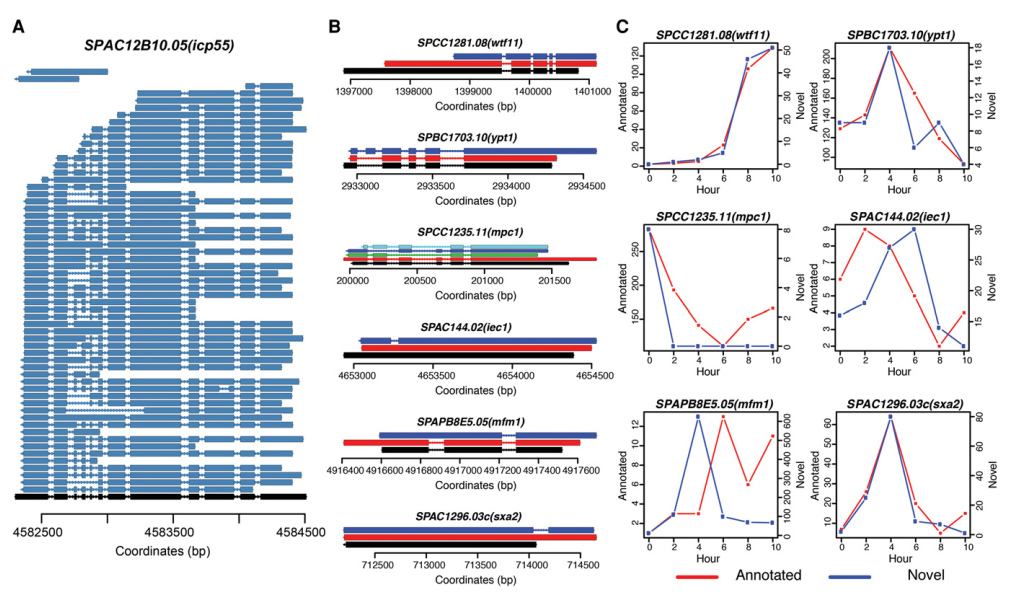
图3 S.pombe减数分裂时期可变剪切事件的实例分析
在S.pombe中主要的可变剪切类型是内含子保留,这可能是由于跳过了个体剪切位点引起的(图4A)。保留的内含子的分布和所有注释内含子的长度分布类似(图4G)。结果发现只有1,300个基因有单独的异构体,1,432个基因有两个异构体,每个异构体至少覆盖一次FL CCS read(图4B)。超过3000个基因有2个以上的异构体,这表明S.pombe中转录组和AS介导的蛋白调控的普遍复杂性。另外,第一次对拥有单一AS事件的异构体(8,739,77.8%)和拥有多重AS事件的异构体(22.2%)进行了区分(图4C)。为了检验AS事件的分析相关性,验证了1708对可变的保留内含子,发现683个极相关的内含子对。相关性用基因间分子关联值表示,相关联的内含子对表现出了较高的关联值,这表示他们的保留比互相排斥更加互相关联(图4J)。尽管在S.pombe中AS是普遍存在的,注释异构体在大多数的基因中是占优势的(3,677个基因有超过90%的reads比对上注释异构体)。但是有648个基因,在已注释异构体占的比例比可变异构体的reads总数更少,(图4D)。大体上,匹配上注释异构体的FL CCS reads几乎比匹配上新异构体的reads要多8倍。除了AS型异构体,我们还发现mRNAs还明显编码770个新TUs和大约3,800个至少覆盖一次read的反义异构体。支持新反义异构体的reads的数量通常比匹配已注释反义异构体的reads数要少很多(图4E)。在已注释的剪切位点中,99.94%的二核苷酸是GU-AG,只有3个例外。但是GU-AG只在新剪切位点中出现了69.67%,或者在新内含子的87.5%的reads中(图4I)。标准剪切位点的新内含子和注释内含子长度相似,但是非常规剪切位点的新内含子大体上更长些(图4H)。将套索测序和PacBio测序进行比较,发现外显子跳跃,新剪切位点和新内含子三种AS事件只有少数是重叠的(图4F),可能是因为两种研究中的条件是独特的,还和捕获不同RNA分子的技术有关。
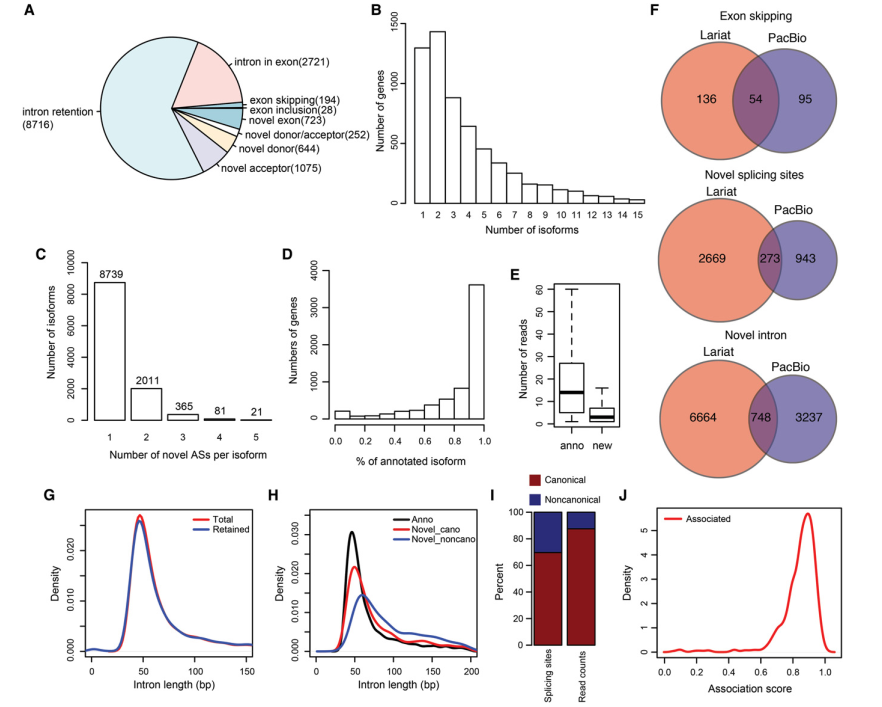
图4 S.pombe减数分裂期可变剪切事件的描述
首先总结了不同AS事件的的总体变化趋势,用每种类型对应的异构体数和reads数表示(图5A)。发现大部分的AS类型在减数分裂期是增加的趋势,但是外显子跳跃事件在减数分裂早期是减少的,在后期却是增加的;内含子保留相对没有变化。下一步,检测基因水平异构体的动态变化。多数个体AS异构体的丰度在减数分裂期是增加的,和总体趋势一致(图5B)。计算基因的注释异构体和可变异构体对应的reads数之间的皮尔森系数(图5C),表明大多数的可变异构体的丰度和注释异构体是相关的。为了检测反相关异构体,挑选了28个超多100个FL CCS reads的覆盖度基因,以做后续分析。新异构体和注释异构体的时间进程模式进行比较,见热图(图5D)。在减数分裂的中期到后期,注释异构体的表达增加,同时发现一个在减数分裂早期表达的48bp内含子保留异构体(图5E,F)。图5E是外显子-内含子结构的实例,图5F是注释异构体和可变异构体的时间进程模式。
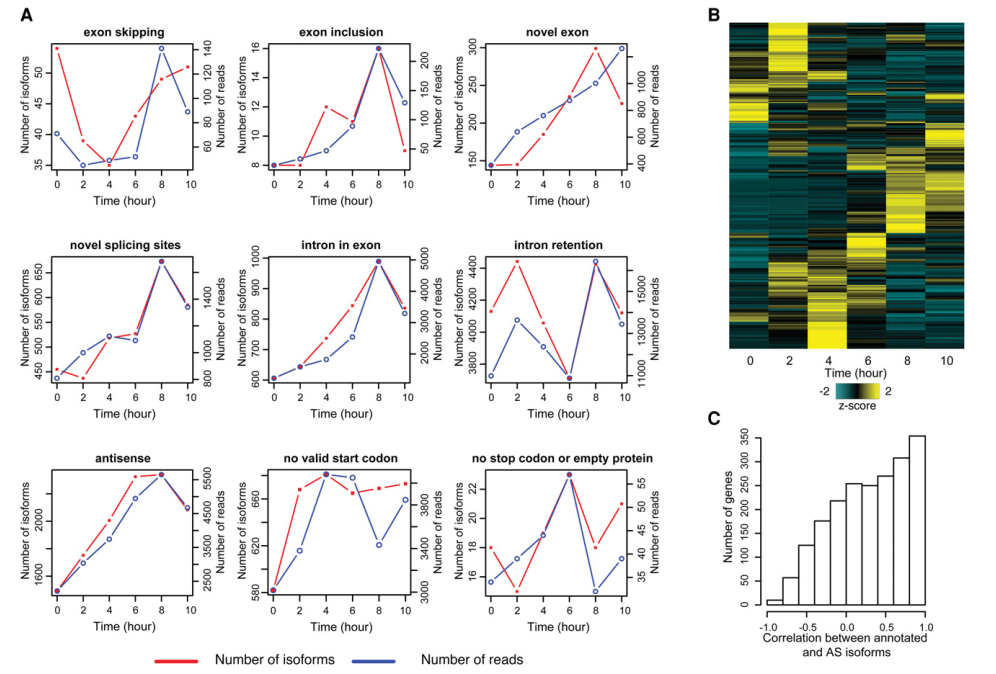
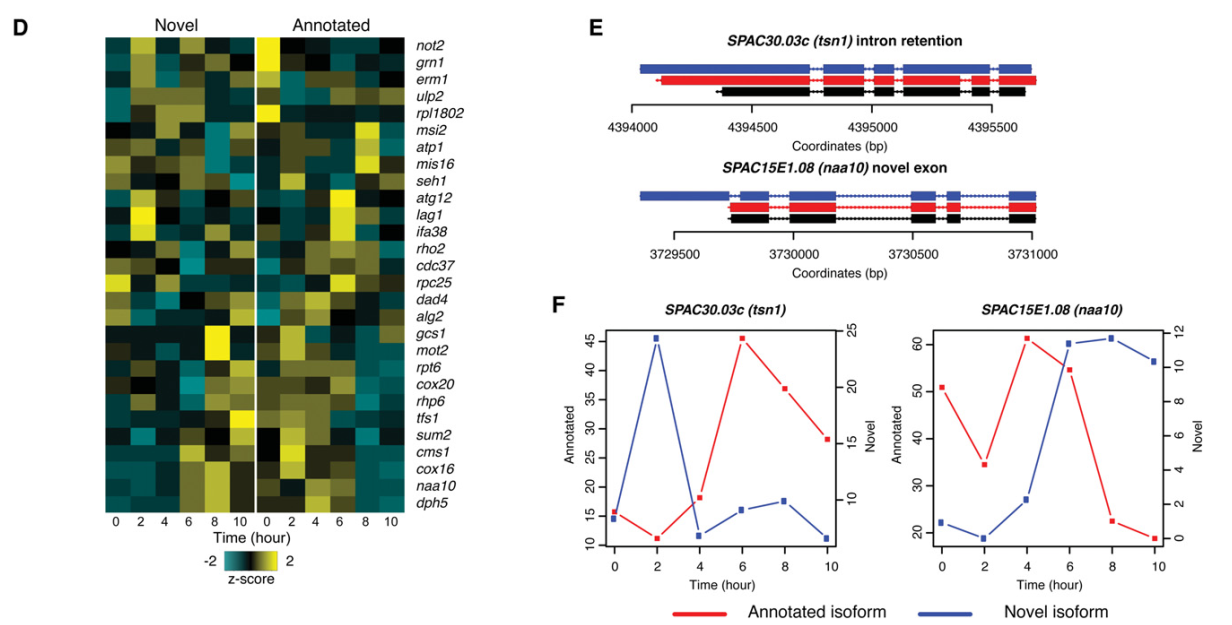
图5 减数分裂期可变剪切的动态变化
可变剪切的主要作用之一就是形成同一基因的独特功能的蛋白质。翻译预测通常是从注释的启动子开始,会在第一个终止密码子停止。测序分析共预测了18,166个异构体是可译的,其他的异构体或是属于非编码蛋白的基因或是缺少终止密码子或是已注释的起始密码子(图6A)。在4,990个基因中,有14,292个独特的可译序列被预测。这些基因中,2,116个基因被预测形成专一的注释蛋白,2,852个基因被预测编码注释蛋白序列和新蛋白序列。还有22个基因编码和注释蛋白序列不同的蛋白序列。大体上,新蛋白序列比注释蛋白序列要短(图6B)。我们发现编码新蛋白序列的异构体的主模式是长度和注释蛋白相似,编码新蛋白序列的异构体次模式是长度小于注释蛋白的20%(图6C)。还有,预测蛋白序列的改变是因为可变剪切通常发生在一个编码序列的开始位置(图6D)。整体上讲,40.3%的带有AS事件的可译异构体编码和注释异构体一样的蛋白。超过半数的其他编码新蛋白序列异构体至少部分和对应的注释异构体是不同的阅读框(图6E)。大约33.9%编码新蛋白序列的异构体和注释异构体是同样的阅读框,其他大约4.07%的异构体随阅读框转移变化而第二改变修复阅读框。47.9%的AS异构体的翻译终止于注释的终止密码子(图6F)。只有2.32%的AS异构体的翻译终止于下游但离注释的终止密码子很近(图6G)。
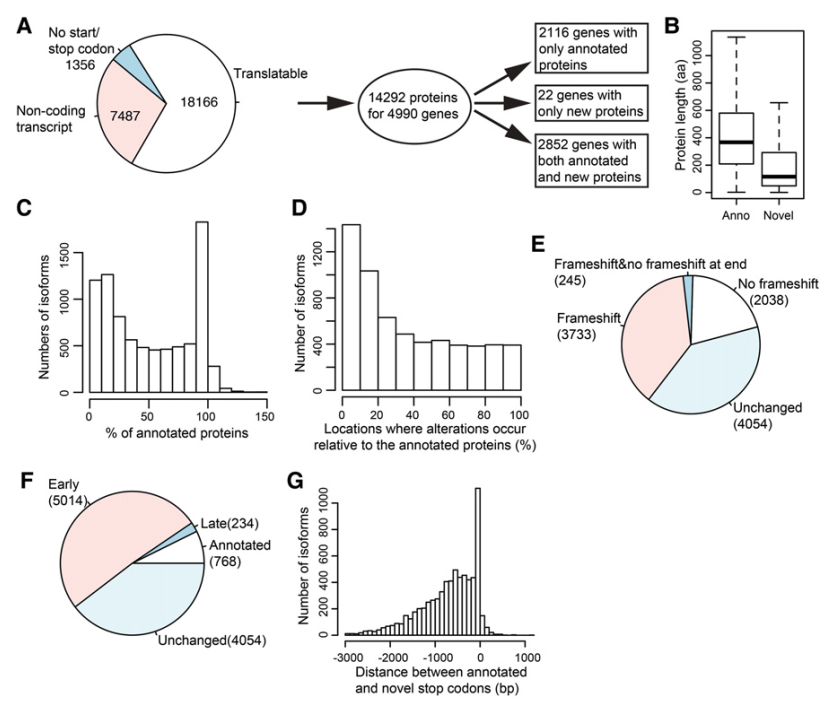
图6 预测检测到的异构体的翻译产物
新序列的保守性通过用BLAST和裂殖酵母,真菌和真核生物比对寻找序列相似的蛋白的方法被评估。二级和三级结构以及他们的特性使用RaptorX预测。对于53个新Tus,大部分和其他裂殖酵母有同源染色体,并没有明显的偏好(图7A)。另外,检测了550个AS异构体的新氨基酸序列的C端的保守性,reads覆盖了>2CCS FL reads,长度至少是19aa。177个AS异构体和其他物种有同源染色体(图7B)。下面,系统评估了新蛋白的二级结构形成。注释蛋白一致表现出低水平的失序,和注释ncRBA的最长ORFs对应的假设蛋白表现出完全的失序(图7C)。研究在插入(内含子保留)和缺失(外显子中内含子)氨基酸序列中的失序残留物的模式,发现低水平失序的主要模式和完全失序的次模式(图7D)。然后,研究AS异构体中可变的C端氨基酸如何影响二级结构。图7E表明大多数改变的C端维持α-helix (H), β-sheet (E). and coil (C)的比例。
图7 保守性和二级结构分析
参考文献:
Kuang Z, Boeke JD, Canzar S. The dynamic landscape of fission yeast meiosis alternative-splice isoforms[J]. Genome Research, 2017, 27(1):145.


















