

2017-08-10
近年来随着二代测序的发展成熟和三代测序的兴起、信息分析方法和工具的发展完善,基于全基因组测序进行动植物群体的进化研究、构建遗传图谱、检测目标性状相关基因已成为常用的策略。应注意到,基因组测序是一个物种研究的起点和基础,更深入的研究通常涉及以下几方面的分析:序列组装、基因组特征、注释效果功能基因和通路研究、进化分析、比较基因组学分析、全基因组复制事件(WGD)、物种起源、驯化、群体遗传等。在这里,我们针对基于de novo测序的植物进化相关研究的最新进展和重要文献作了简要的梳理,以便为项目提供参考。
1. 植物基因组denovo测序技术概述
基因组从头测序(de novo sequencing)是指对基因组序列未知或没有近缘物种基因组的某个物种的全基因组序列的测序。不需要参考资料,测序后用生物信息学手段对测序序列进行拼接、组装和注释,从而获得该物种的基因组序列图谱。目前常用的测序平台包括二代和三代,如Illumina HiSeq、Illumina MiSeq、PacBio RS、PacBio Sequel等。
植物基因组通常是多倍体,基因组大,杂合度高,具有高度重复序列和全部或部分的基因组重复片段,这些特点造成了其基因组测序组装的难度。目前已发表的植物基因组大多是基于短读长测序组装,结果碎片化。对此,三代测序技术,即PacBio单分子实时(SMRT)测序技术的出现有助于可以解决这一问题。三代测序技术解决了二代测序高GC区域无法准确测定、高重复序列无法跨越、海量短序列组装困难等几大困扰,超长的读长不仅给GC含量异常和高重复序列基因组组装提供了很好的契机,而且能够大幅度提高已有基因组的组装指标。目前,三代结合二代测序技术是基因组从头测序的选择,大规模地物种全基因组denovo测序已渐入佳境。
测序流程如下:
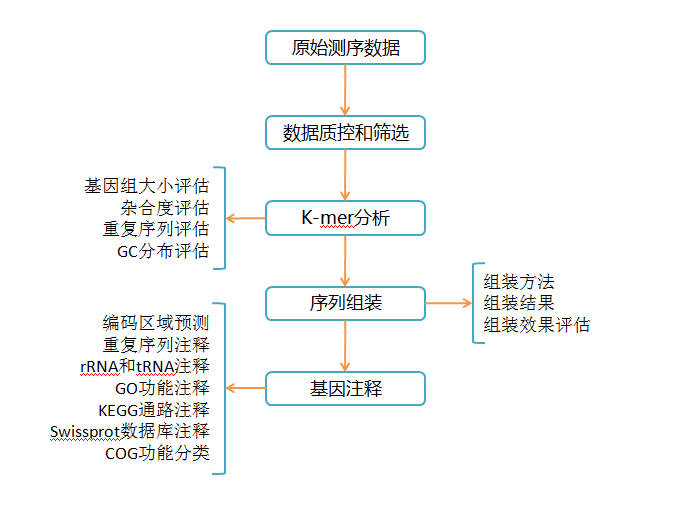
2. 数据组装
高等植物基因组常为高度杂合,为测序数据的组装带来一定挑战。针对这一问题,常用的研究方法是通过选择双单倍体或构建高纯合度的自交系进行测序来降低组装难度。
2.1 组装方法
常用组装软件有MaSuRCA、SOAPdenovo2、Opera、Platanus、SSPACE、GapCloser等。
其中,MaSuRCA软件可对短序列和长序列联合分析,通过产生superreads提高计算效率和容错度。SOAPdenovo2软件适用于大基因组,可减少构图过程中的内存消耗,提高gap覆盖度。SSPACE软件特点是运行时间短,可实现双端测序数据集的多重文库输入和contig延长。
2.2 组装结果
组装结果常用contig N50、scaffold N50、superscaffold、基因组覆盖度等指标代表。
2.3 组装效果评估
常用评估方法有:CEGMA、EST、BAC、RNA-Seq、EST+RNA-Seq等。
3. 基于Denovo测序的高等植物进化研究进展
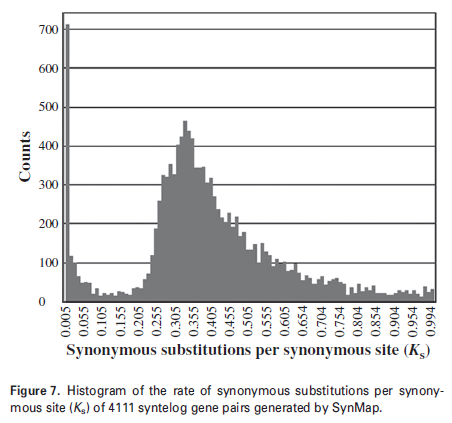
3.1 核桃全基因组倍增事件验证(The Plant Journal,2016年9月)
化石证据表明胡桃属WGD发生于60百万年前(Mya),利用核桃全基因组测序结果,采用自我比对的方法,鉴定基因组部分同源序列间的共线性保守区,识别到8459对旁系同源基因,其中4111对相关基因涉及转录调控蛋白和信号传导蛋白的编码,对这些基因(Ks<1)构建Ks直方图,图中主峰位置在Ks=0.33。研究结果与14对旁系同源基因的分歧时间一致(Ks = 0.274±0.09) (Luo et al., 2015),为WGD提供了有力支持。但还需通过种间比较精确确定系统发育的时间。
3.2 茄科植物进化与辣椒驯化(PNAS,2014年8月)
采用OrthoMCL方法进行基因家族的种间比较(辣椒、番茄、马铃薯、拟南芥)构建单拷贝同源基因的系统进化树,发现在36Mya辣椒与番茄、马铃薯分离,即茄科辣椒属形成,期间发生了辣椒染色体易位、倒位等变异,156Mya茄科出现,紧随着单、双子叶植物的分离。与葡萄比较发现了辣椒基因组三倍化,这可能是茄科的共同事件,但三倍化后出现了基因拷贝的丢失。通过4DTv方法计算WGD时间,WGD峰出现在0.3位置。
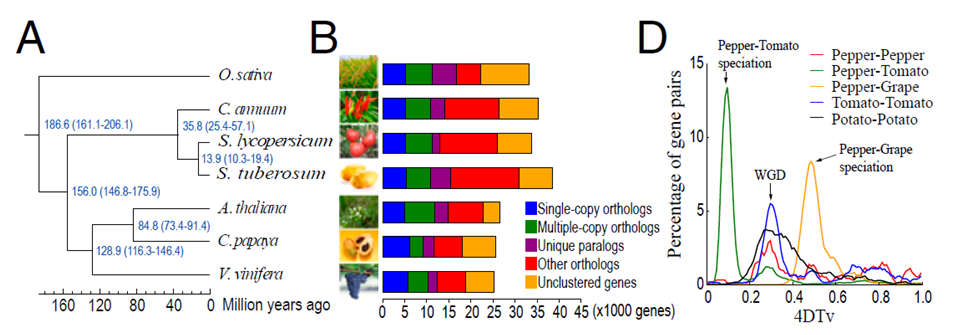
辣椒驯化研究选择了18个栽培品种和2个野生/半野生品种,通过遗传瓶颈法鉴定人工选择标记,通过θπ、θω值检测遗传多样性降低鉴定了115个人工选择区域(含511个基因),其多态性水平显著降低,相关基因功能涉及转录调节、胁迫与防御响应、蛋白-DNA复合物装配、生长和果实发育等,与栽培种与野生种的形态和生理差异相关。

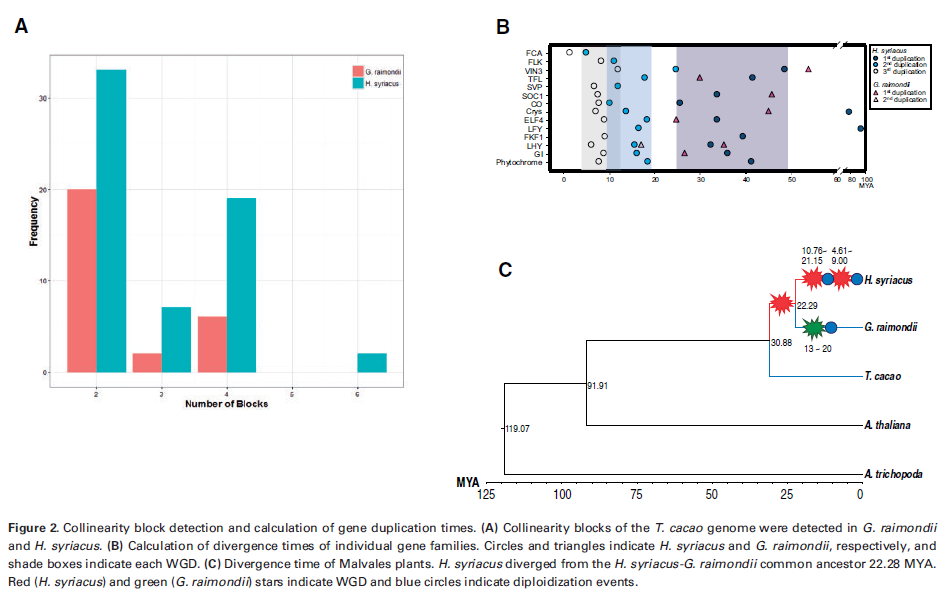
3.3 锦葵科植物基因组结构与多倍化(DNA Research,2017年2月)
MCScanX检测共线性模块,以可可树基因组为模板,检测木槿和雷蒙德氏棉的共线性模块,发现木槿共线性模块的数目是雷蒙德氏棉的4倍,大小为其2倍,表明木槿中出现过WGD。系统发育分析揭示基因组复制模式,GI、CONSTANS和SOC1等基因复制表明木槿中出现了3次WGD,但很多基因在第一次复制后出现丢失。
通过BEACT构建系统发育树,计算Ks并估算锦葵科分离时间,结果表明91.1Mya锦葵科从十字花科-锦葵科共同祖先分化出来,木槿在物种形成前、物种形成后25.23~48.23Mya和4.61~21.15Mya的时间内分别出现3次WGD,WGD与随后的二倍化导致基因量不均衡调节和基因家族CNV。
3.4 甜橙基因组进化(Nature Genetics,2013年1月)
旁系同源基因家族的累积大小和频率可作为WGD的标记,通过自我比对鉴定了1296个旁系同源基因,复制基因的平均Ks值表明远古WGD事件,没有近期WGD。进一步基于种间共线性模块,估算了至少49个染色体易位与融合为双子叶植物共有,系统发育显示甜橙、可可树、拟南芥和番木瓜近缘,柑橘属在85Mya从锦葵目分离。
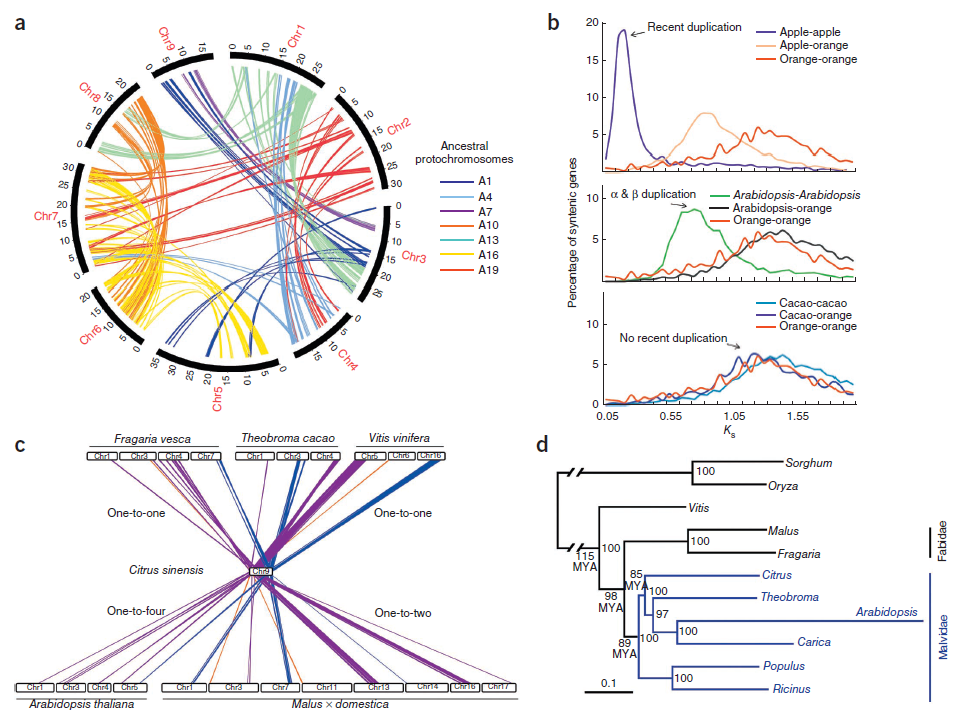
杂交和多倍化是植物最重要的进化方式之一。总结以上文章的研究思路,我们可以看到通常一个植物基因组项目进化分析的关注点包括全基因组复制事件、同源比对以研究古多倍化、自身比对以研究近多倍化、系统发育树构建以研究群体聚类和起源等。对于具体项目而言,可根据关心的问题和物种特性选择相应的分析方法。
参考文献
[1] Martínez-García P J, Crepeau M W, Puiu D, et al. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols[J]. Plant Journal for Cell & Molecular Biology, 2016, 87(5):507-532.
[2] Qin C, Yu C, Shen Y, et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization.[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(14):5135-40.
[3] Kim Y M, Kim S, Koo N, et al. Genome analysis of Hibiscus syriacus provides insights of polyploidization and indeterminate flowering in woody plants[J]. Dna Research An International Journal for Rapid Publication of Reports on Genes & Genomes, 2017, 24(1):71-80.
[4] Xu Q, Chen L L, Ruan X, et al. The draft genome of sweet orange (Citrus sinensis).[J]. Nature Genetics, 2013, 45(1):59-66.


















