

2017-06-28
BSA(Bulk segregant analysis)称为混合分组分析法,是近年来兴起的一种快速简单的性状定位的方法。广泛运用于拟南芥、水稻、玉米、黄瓜等物种上。BSA的特点之一就是将两种极端性状的子代个体混合测序(pool-seq)。但BSA绝对不是混池测序这么简单,实际上根据群体材料、实验设计的不同,BSA又有各种不同的方法。接下来就为大家一一介绍。
QTL-seq
QTL即数量性状位点,在定位QTL时需要挑选两个性状差异较大(抗病/感病、高杆/矮杆)的纯合亲本进行杂交,构建F2群体或RIL群体。由于研究的数量性状,子代表型不会非此即彼,而是在群体中呈连续的正态分布。选择表现出极端性状的20~30个个体进行混池测序,比如极端感病和极端抗病、最高和最矮的个体。我们通常选择要研究的性状的亲本作为参考序列,将混池测序的reads比对到参考序列,计算SNP-Index。因为基因遗传的随机性,大部分SNP-index会在0.5附近。但与所研究性状相关QTL区域,两个混池间SNP-index差异会非常大,通过这一原理我们可以将性状相关QTL大致定位到基因组上某一区域。

MutMap
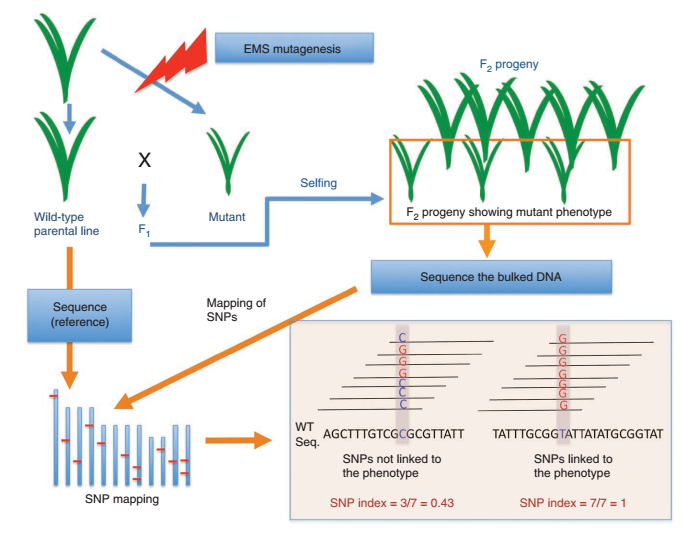
当面对通过EMS诱变产生的突变性状时,我们采取了与QTL不同的方法,称之为MutMap即Mutation+Map。该方法先将突变体与野生型亲本进行杂交,得到F1代。F1代再自交得到F2代,在F2代中选择突变型个体进行混池测序。然后将测序得到的reads比对到亲本基因组上,计算SNP-index。由于突变体为纯合个体,性状相关SNP-index应该为1,由于连锁关系,相邻位点SNP-index也会增加。其余无关SNP位点的SNP-index为0.5。因此SNP-index图会出现一个峰值,性状相关候选基因就位于该区域内。Mutmap的方法杂交时间短,相对于QTL定位更加省时高效;由于突变个体纯合,背景更纯,噪音少;只用对突变体混池和野生型亲本进行测序,成本更低,分析更简单。
Mutmap+
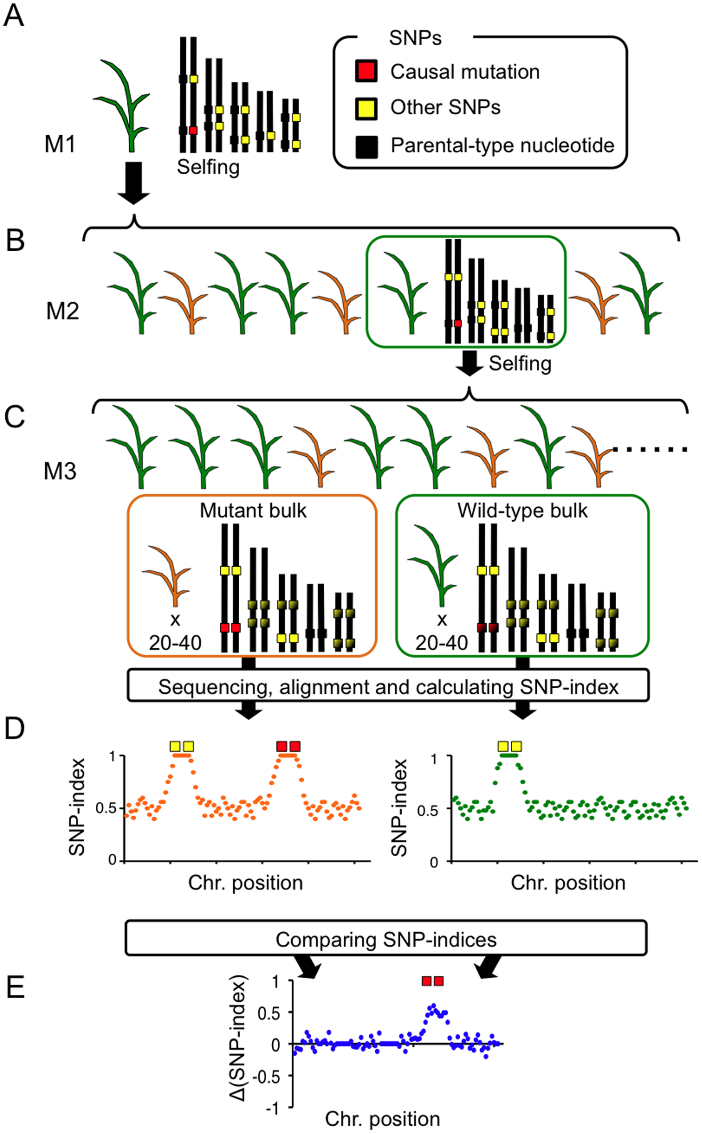
Mutmap在针对EMS诱导突变的性状研究中优势非常明显,但并不是所有性状都适合用Mutmap的方法。对于纯合突变致死、不育或者严格自交且人工杂交困难的物种就无法使用Mutmap的方法进行研究。莫慌!有新的方法可以应对,我们称之为Mutmap+。该方法是将EMS诱导产生的突变体M1自交产生M2,选择M2中含有突变基因的野生型个体进行自交产生M3。M3发生性状分离,选择M3子代中野生型和突变型各20株个体,混池测序。将reads分别比对到亲本的基因组上,计算SNP-index。由于M2许多突变位点已经纯合,M3上很多区域SNP-index为1。但与突变位点相关的SNP-index为1是只有突变池才有的。所以计算△SNP-index,去除背景噪音,就可以找到突变所在基因区域,定位突变基因。
Mutmap-gap
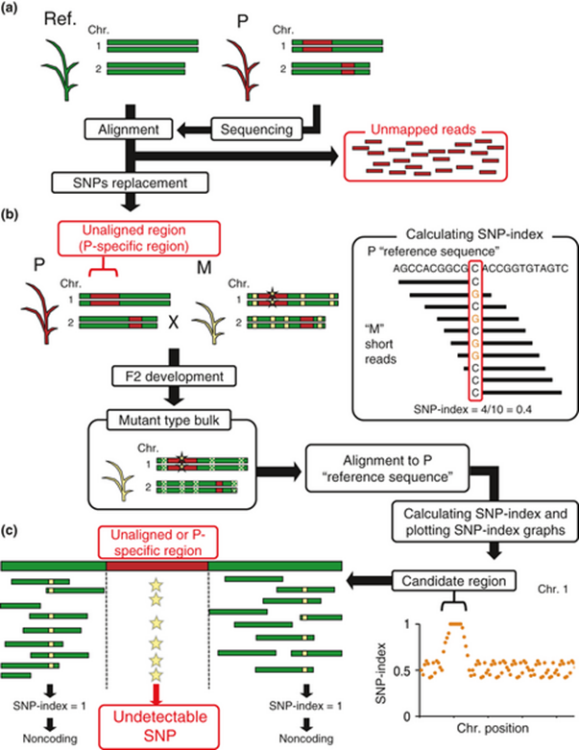
对于以重测序为基础的分析内容,都需要有参考基因组。但往往研究的样本与数据库中的参考基因组属于不同品系,因此基因组上存在一定的差异。如果EMS诱导的突变发生在一个参考基因组没有的基因上,那么用上述的MutMap和MutMap+就无法找出该候选基因。而MutMap-Gap的方法就是结合了MutMap和de novo组装的一种基因定位方法,从而解决参考基因缺失位点上的基因突变问题。
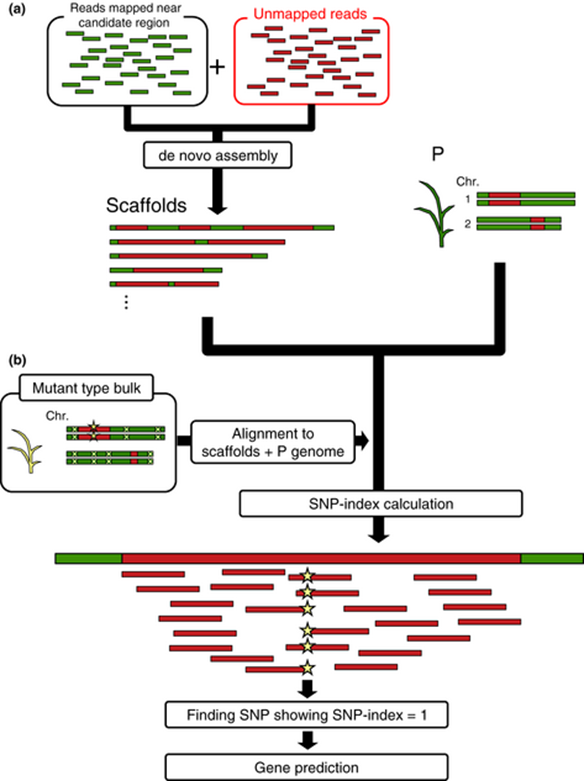
Mutmap-Gap首先将所研究品系的野生型和参考基因组比对,得到所研究品系的参考基因组。然后利用EMS诱变,再进行MutMap分析,得到SNP-index图。对位于SNP-index图peak区域内的基因进行分析,如果在这个区域内找不到跟突变性状相关的基因,那么突变位点很有可能就在品系特有基因区域内。这时候我们将之前比对不上参考基因组的野生型亲本unmapped reads,和位于这个peak区域的野生型亲本mapped reads一起进行de novo组装,得到潜在的新基因,再以此为参考计算SNP-index。
总结:
这四种方法都是基于纯合基因型亲本的,都是通过将杂交家系中具有极端性状个体混合测序,计算SNP-index,来实现基因定位。不同点在于需要测序的样本以及其适用范围。QTL-seq需要测序的为两个亲本和两个极端性状子代混池,适用于质量性状、有明显主效基因的数量性状;Mutmap需要测序的样本为突变型子代混池和野生型亲本,适用于诱变突变性状;Mutpmap+需要测序的样本包括野生型亲本、突变型子代池和野生型子代池,适用于早期致死突变或无法异交的突变株;Mutmap-Gap适用于目标基因不在物种参考基因组上的性状。
但总的来讲,相对于其他的性状定位方法,利用BSA技术进行性状定位周期更短,价格更低,定位效果更好。
参考文献
[1] Takagi H, Abe A, Yoshida K, Kosugi S et al. 2013. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. The Plant Journal (2013) 74, 174–183
[2] Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M et al. 2012. Genome sequencing reveals agronomically important loci in rice using MutMap. Nature Biotechnology 30: 174–178.
[3] Fekih R, Takagi H, Tamiru M, Abe A, Natsume S, et al. 2013 MutMap+: Genetic Mapping and Mutant Identification without Crossing in Rice. PLOS ONE 8(7): e68529.
[4] Takagi H1, Uemura A, Yaegashi H, Tamiru M, Abe A, Mitsuoka C, Utsushi H, Natsume et al. 2013. MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii.New phytologist New Phytologist 2013 200(1):276–283.


















