

2017-06-01
与二代测序技术相比,三代测序技术具有读长长、无GC偏好性的特点。虽然目前三代测序技术单条测序序列的错误率高于二代测序技术,但是由于这些错误都是随机的,因此可以通过提高测序深度来有效地减少错误率。三代测序技术的这些特点,使其在基因组从头组装方面大显身手,能够有效地提高组装序列contig N50的长度。但是想要得到染色体级别的基因组组装序列,光靠三代测序技术还是不够的,还需要借助其他的技术手段。比较常用的手段有遗传图谱、长插入片段文库以及光学图谱等技术手段。
长插入片段文库能够跨过较大的重复片段的区域,因而在以往的基因组组装中都发挥着重要作用。与BAC库等长插入片段文库相比,fosmid库具有构建时间短、花费较低、基因组覆盖均匀等特点。将这些长片段文库再构建小的subclone,然后利用一代测序技术测序能够获得精确的基因组信息(clone-based 组装策略)。目前很多模式生物的基因组都是基于这种策略来构建的,比如人,水稻,拟南芥等。但是这种策略构建基因组序列费用非常高。直接对这种长片段文库的两端测序,获得的序列能够用于构建scaffold,提高全基因组鸟枪法测序策略的组装片段长度。新兴的光学图谱技术根据内切酶酶切位点的信息,能够提供几百Kb到几Mb范围的基因组物理图谱。此方法获得的光学图谱也可以用于构建scaffold,与fosmid库相比,更加快速,而且花费低。但是由于短的contig(<100 Kb)上缺少酶切位点,因而使用此种方法无法锚定到scaffold上。遗传图谱则能够将contig/scaffold锚定到linkage group上,使组装水平达到染色体级别。需要注意的是,如果contig/scaffold组装的效果不好,加上遗传图谱,虽然可以得到染色体级别的基因组组装水平,但是序列中会含有大量的N。基于这种基因组序列的基因注释水平等都比较低,不利于后续功能研究。
今年5月,Nature communications在线发表了一个迄今为止最高质量的植物基因组序列。该研究组装的物种是水稻(Oryza sativa, 2n=2x=24),品种是Shuhui498,组装策略综合利用了三代单分子测序技术、fosmid文库和Bionano光学图谱,最终组装的序列仅有1%的缺失。水稻现有的参考基因组为日本晴(Nipponbare),是基于clone-based的策略构建的。Shuhui498最终组装的准确性和完整性都优于日本晴。下面小编就来分享一下这篇文章,看如果利用这些技术得到了如此高质量的基因组序列。
测序策略
本研究获得了47 Gb的PacBio的序列,覆盖基因组~118 X。同时构建了插入片段大小为~40 Kb 的fosmid文库,564个fosmid pools,每个pools有~1,000个clone。对这些fosmid文库,本研究采用了GBS的测序方法,每个tag的测序深度为~3 X,共获得6.3 Gb的数据。本研究还构建了Shuhui498和Nipponbare的重组自交系。为了构建遗传图谱,作者挑选了F3群体中364个个体,采用GBS测序的方法,每个tag的测序深度为~4 X,每个个体平均测序数据为73 Mb,一共获得26.9 Gb的数据。本研究获得了99 Gb的光学图谱数据,基因组覆盖度为250 X。构建这些光学图谱的分子片段>100 Kb, N50为202 Kb。这些数据组装后共得到453个genome map,总长度为406 Mb,N50为2.48 Mb。此外,本研究也构建了插入片段为450 bp的二代测序文库并获得了38.7 GB的测序数据,覆盖基因组~100 X。本研究还对多个组织进行了RNA-seq,用于更好的注释基因组和评估组装效果。具体的测序策略见下表。
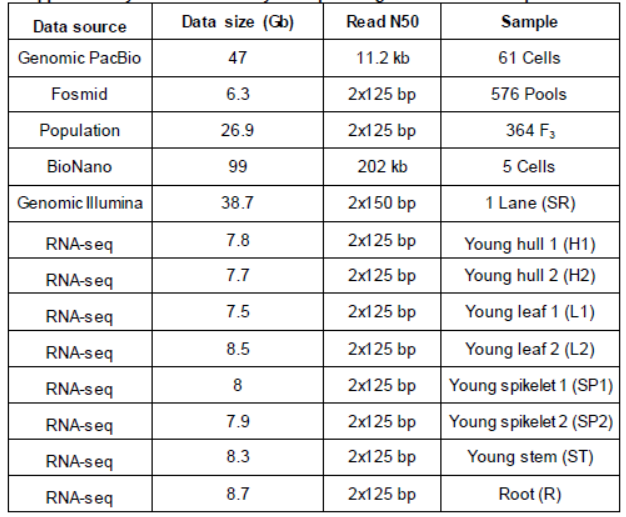
测序的策略
组装策略
1. Contig构建
首先,采用PBcR pipeline对原始的PacBio序列纠错,获得16.2 Gb的纠错后序列。然后采用PBcR的low stringency (LS) 和high stringency (HS)模式、CANU和Falcon组装纠错后序列。同时,将fosmid测序的序列比对到纠错后的序列上,挑选出含有fosmid测序序列的PacBio序列,并对每个fosmid pool的序列单独组装,获得fosmid contig。contig组装的详细结果见下表:
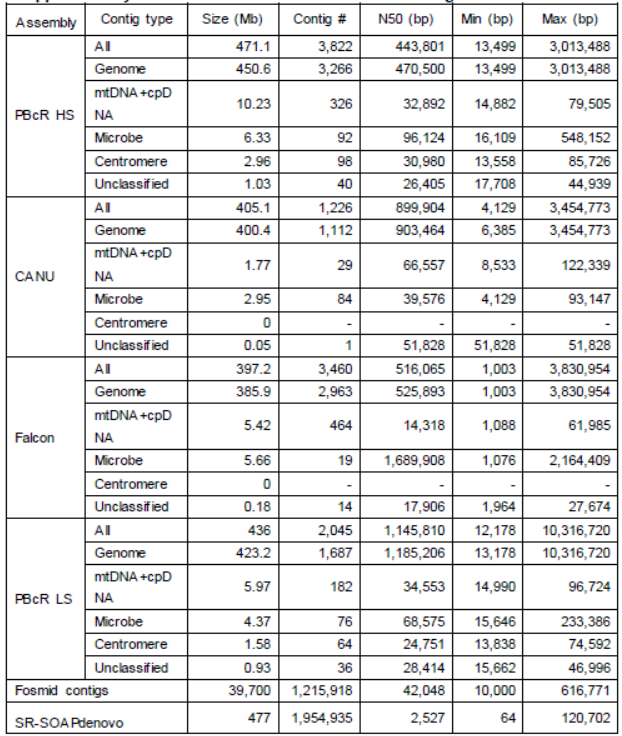
Contig组装的结果
2. 遗传图谱构建并将contig锚定到遗传图谱上
PBcR LS组装的contig N50最长,因此被选为reference来构建遗传图谱。该遗传图谱共获得12个linkage group (LG)。497个contig锚定到了该遗传图谱上,序列总长为355.9 Mb。
3. Super-contig构建
被锚定到遗传图谱上的497个contig,被进一步连成super-contig。原理就是如果fosmid contig与两个contig有overlap,那么这两个contig就被连成一个super-contig。全基因组组装的时候,也会产生一些错误的contig。这些contig也能够在super-contig构建的过程中被纠正。原理就是如果一个contig与周围的contig有overlap,而且这个overlap也被fosmid contig验证,那么这个contig就会在有overlap的地方分开,分成多个contig。具体的原理图见下图:
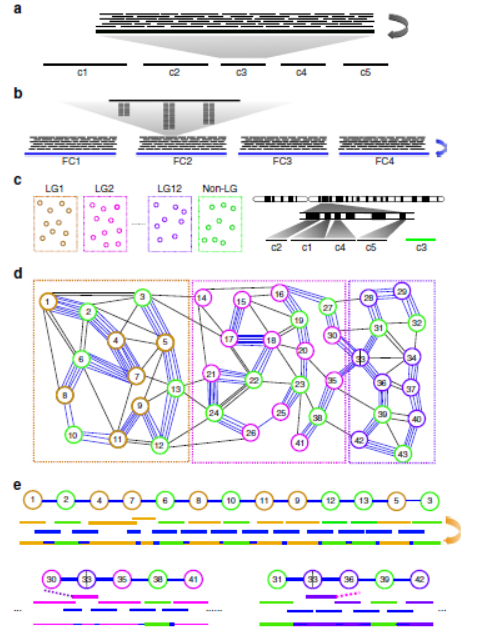
Super-contig构建的原理
4. Super-contig纠错
Bionano构建的genome map被用于super-contig的纠错。作者首先查看了着丝粒和近端粒区域,发现着丝粒区域与genome map完全一致,而24个近端粒区域中有21个与genome map一致。然后Quiver被用于来纠正≤2 Kb的组装错误。
5. 组装效果评估
将Illumina DNA的短序列,RNA-seq的数据比对到组装的基因组上,评估出基因组组装的碱基错误率<0.0017%。这个错误率低于水稻现有的参考基因组日本晴的序列。而存在于最终组装序列的fosmid contig的碱基错误率为0.0017%,略高于其它区域,可能是以为这些区域含有高重复序列。基于这个比对结果和genome map的比对结果,大约有1%的序列未组装到最终的基因组序列中。
Shuhui498与日本晴基因组比较
比较Shuhui498与日本晴基因组序列,发现Shuhui498的着丝粒区域更完整、含有更少的叶绿体和线粒体序列。并且,二者在染色体上的相似性非常高,不过也存在~250万个SNP和很多的大片段结构变异(具体见下图)。比较二者的基因序列发现,二者只有~27%的基因蛋白质序列完全一致。虽然二者的重复序列比例一致,但是重复序列的内容却不一样。

Shuhui498与其它水稻基因组比较
最后,本研究比较了Shuhui498基因组和其它17个 高覆盖测序的水稻基因组序列。Shuhui498与同属于籼稻的MH63 和 ZS97基因组更相似。同时,本研究鉴定了Shuhui498和日本晴基因组中相对于其它水稻基因组的presence variation (PV),发现PV广泛存在。
总结
利用三代单分子测序技术,再结合长插入片段文库、光学图谱和遗传图谱数据,本研究构建了一个仅有1%缺失的基因组序列,是迄今为止质量最高的植物基因组。
参考文献:
Du H, et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat Commun. 2017, 8:15324. doi: 10.1038/ncomms15324.


















