

2025-02-14

文章题目:Comprehensive multi‐tissue epigenome atlas in sheep: A resource for complex traits, domestication, and breeding
兰州大学王维民教授团队在iMeta在线发表了题为“Comprehensive multi-tissue epigenome atlas in sheep: A resource for complex traits, domestication and breeding”的文章。研究通过整合多组学数据集,为未来研究绵羊复杂性状提供了基础资源和成功案例。本研究中转录组检测及部分数据分析工作由上海派森诺生物科技股份有限公司完成。
一、研究亮点
✴基于9个重要组织的92个转录组和表观基因组数据集,构建了首张绵羊多组织表观遗传调控图谱;
✴鉴定出753723个非冗余功能元件,其中60%以上为首次发现,主要包括与感知能力、免疫反应和尾部脂肪沉积相关的组织特异性启动子和增强子;
✴利用多维组学数据集鉴定出一个新的变异,Chr13:51760995A>C,位于激活增强子区域,影响绵羊尾部脂肪沉积。
二、研究背景
绵羊(Ovis aries)作为重要的农业动物之一,具有丰富的经济价值,但对其适应性进化和重要农艺性状的分子遗传机制仍知之甚少。此外,全面的功能基因组注释对于阐明畜禽农艺性状的分子机制至关重要,但目前仍缺乏对绵羊基因组功能元件的系统注释。
三、研究方法
研究材料:两只6月龄半同胞湖羊公羔的18个组织样本(软骨、盲肠、垂体、下丘脑、肝脏、背最长肌、瘤胃、脾脏和尾脂)
研究方法:RNA-Seq、ATAC-Seq、CUT&Tag和Hi-C
四、研究路线
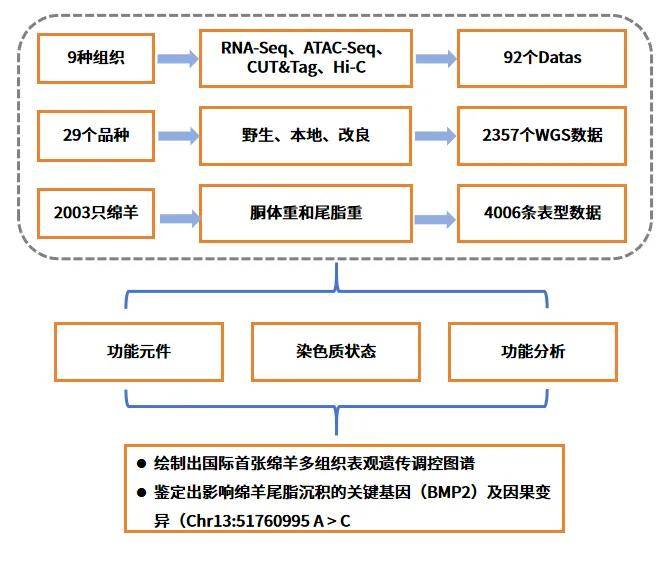
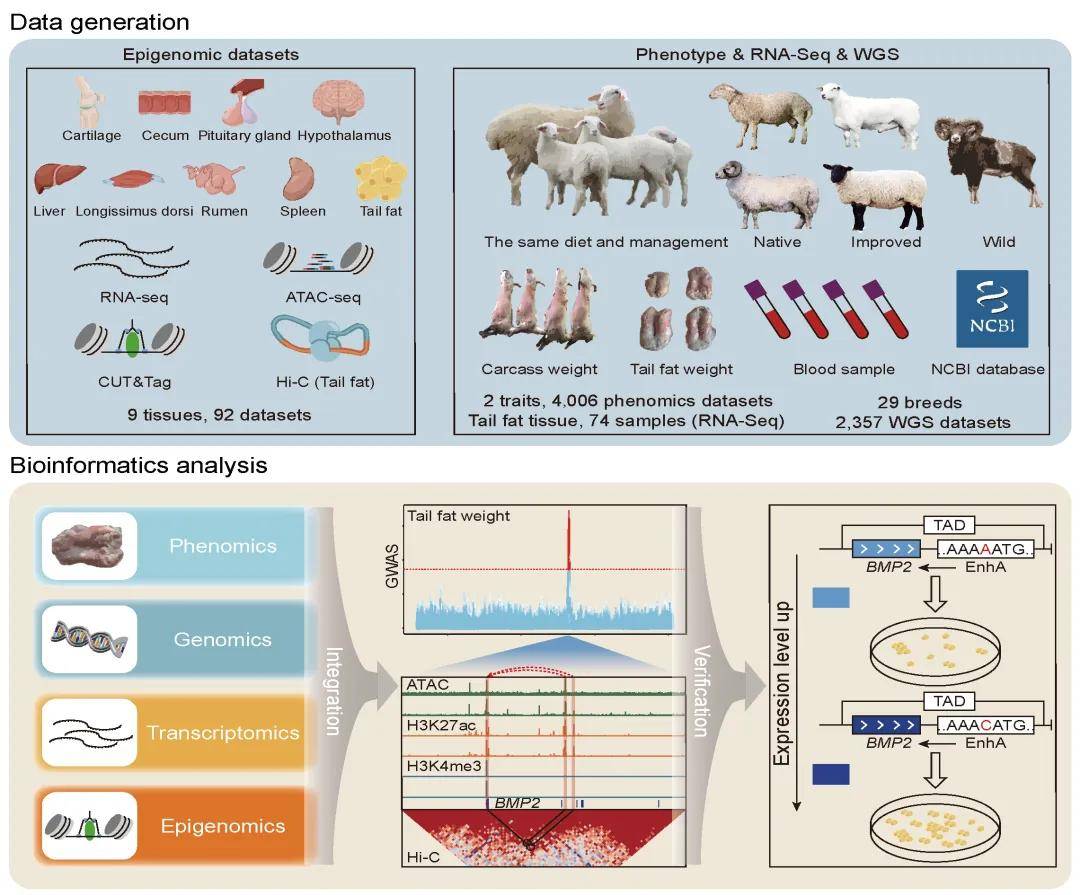
五、研究结果
1、绵羊多组织表观基因组图谱概述
为推进绵羊基因组的功能注释和探讨增强基因组注释对解释绵羊复杂性状的作用,研究小组对9个重要组织(软骨、盲肠、垂体、下丘脑、肝脏、背最长肌、瘤胃、脾脏和尾脂)进行了RNA-Seq、ATAC-seq、两种组蛋白修饰(H3K27ac和H3K4me3)的CUT&Tag和Hi-C测序,生成了92个表观基因组数据集。为探讨增强基因组注释在解释绵羊复杂性状中的作用,整合了全球29个品种的364个全基因组测序(WGS)数据,用于群体结构和选择信号分析。同时收集了2,003只绵羊的胴体重和尾脂重表型数据以及WGS数据。从ATAC-seq、H3K27ac和H3K4me3数据中分别鉴定出258403、20945和52552个peaks,ATAC peaks被注释到非编码区域,而H3K27ac和H3K4me3 peaks主要被注释到基因的启动子区域。此外,共鉴定出753723个非冗余调控元件,包括40606个潜在的启动子、158677个潜在的增强子和846058个开放染色质区域,其中超过84%和64%的增强子和启动子是本研究中新鉴定的,50.51%的启动子和31.35%的增强子与已发表肝脏数据中鉴定的重叠。
相关性结果显示,ATAC-seq、H3K4me3和H3K27ac数据集彼此之间表现出很强的正相关性(平均R = 0.68),三种表观遗传标记信号分布在基因转录起始位点(TSS)周围的区域。以两只绵羊的尾脂组织为代表性组织进行了原位Hi-C测序,并在25kb的分辨率下构建了全基因组互作图谱。结果发现,染色体之间的空间距离越近,相互作用程度越高,尤其是在同一个染色体中。基于上述数据集生成了高分辨率、多组织的表观基因组图谱,丰富了绵羊基因组中功能元件的注释。
 图1 绵羊多组织表观基因组图谱
图1 绵羊多组织表观基因组图谱
2、跨多组织和物种染色质状态的预测和表征
整合9种组织中的三个表观遗传标记数据集(ATAC-seq、H3K4me3和H3K27ac),共定义六种染色质状态,主要分为启动子(TssA和TssW)、增强子(EnhA和EnhAW)、ATAC岛和静止/抑制区域。启动子状态(TssA和TssW)在TSS处表现出最高的富集度,并且与其他染色质状态相比,其基因表达平均水平最高。此外,利用公开的DNA甲基化数据研究了组织中DNA甲基化和染色质状态之间的关系。结果发现激活启动子的甲基化水平最低,这证实了众所周知的基因表达和启动子甲基化水平之间的负相关性。全基因组染色质状态图谱显示抑制区域分布在整个基因组的大部分区域,除此之外其他染色质状态的分布相似。还检查了不同组织中染色质状态的分布,结果表明,启动子状态(TssA和TssW)的活性变化较小,而增强子状态(EnhA和EnhAW)的活性在组织之间变化较大。
比较绵羊、牛和猪基因组之间激活启动子(TssA)和强激活增强子(EnhA)序列的保守性。结果显示,绵羊基因组中80.45%的TssA和81.99%的EnhA区域在牛基因组中保守,而猪基因组中只有25.87%的TssA和30.66%的EnhA保守。同时,在绵羊、牛和猪的同源基因CD86附近均观察到保守和非保守的TssA和EnhA区域,其中H3K4me3和H3K27ac组蛋白修饰模式在这三个物种中是一致的。这表明调控元件在进化关系更近的物种之间往往更保守,而高度保守的调控元件可能在不同物种之间发挥相似的功能作用。
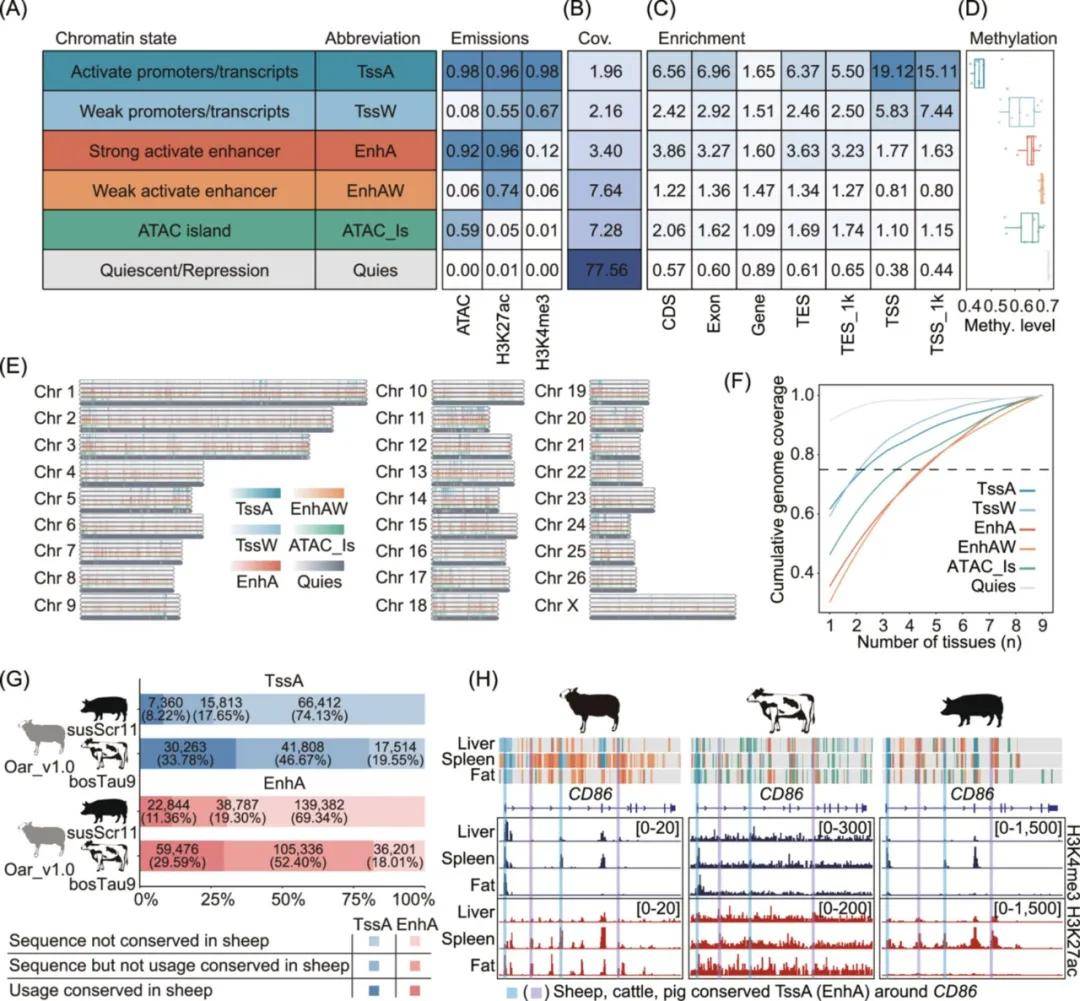 图2 九种组织的全基因组染色质状态图
图2 九种组织的全基因组染色质状态图
3、基因和染色质状态的组织特异性分析
在9种组织中总共检测到8334个组织特异性基因。这些组织特异性基因的功能预测准确反映了各种组织的特定生物学功能。基于qRT-PCR对组织特异性基因中代表基因的表达进行检测,验证了分析结果的准确性。此外与其他组织相比,组织特异性基因在相应组织中表现出更强的活跃染色质状态富集和较少的抑制区域。同时,根据基因重叠EnhA的数量将基因分为三组。结果表明,与EnhA区域重叠较少的基因相比,EnhA区域重叠较多的基因表达水平较高,组织特异性较低。GO富集分析表明,只有一个EnhA重叠的基因与组织特异性功能相关,例如脾脏中的溶酶体活性和吞噬作用。相反,具有多个EnhA重叠区域的基因富集了更普遍的生物学功能。
在九种组织中鉴定了184019个组织特异性EnhA,发现组织特异性EnhA区域信号与组织特异性基因表达之间存在显著正相关(p<0.05)。GO富集分析结果表明,所有与组织特异性EnhA重叠的基因都表现出相应组织的已知生物学功能。同时还对组织特异性EnhA序列中的motif进行了富集分析,发现与其他组织相比,相应组织中的motif对应转录因子表达水平更高。这表明组织特异性EnhA区域可能通过组织特异性转录因子(TF)与组织特异性motif之间的结合来调控基因,从而影响基因的组织特异性表达。此外,激活启动子也表现出组织特异性功能,尽管与EnhA相比程度较低,但进一步支持了活性染色质状态在调节组织特异性功能中的关键作用。结果表明,染色质状态的系统分析对下游整合表观基因组数据以解析绵羊农艺性状变异的遗传机制非常重要。
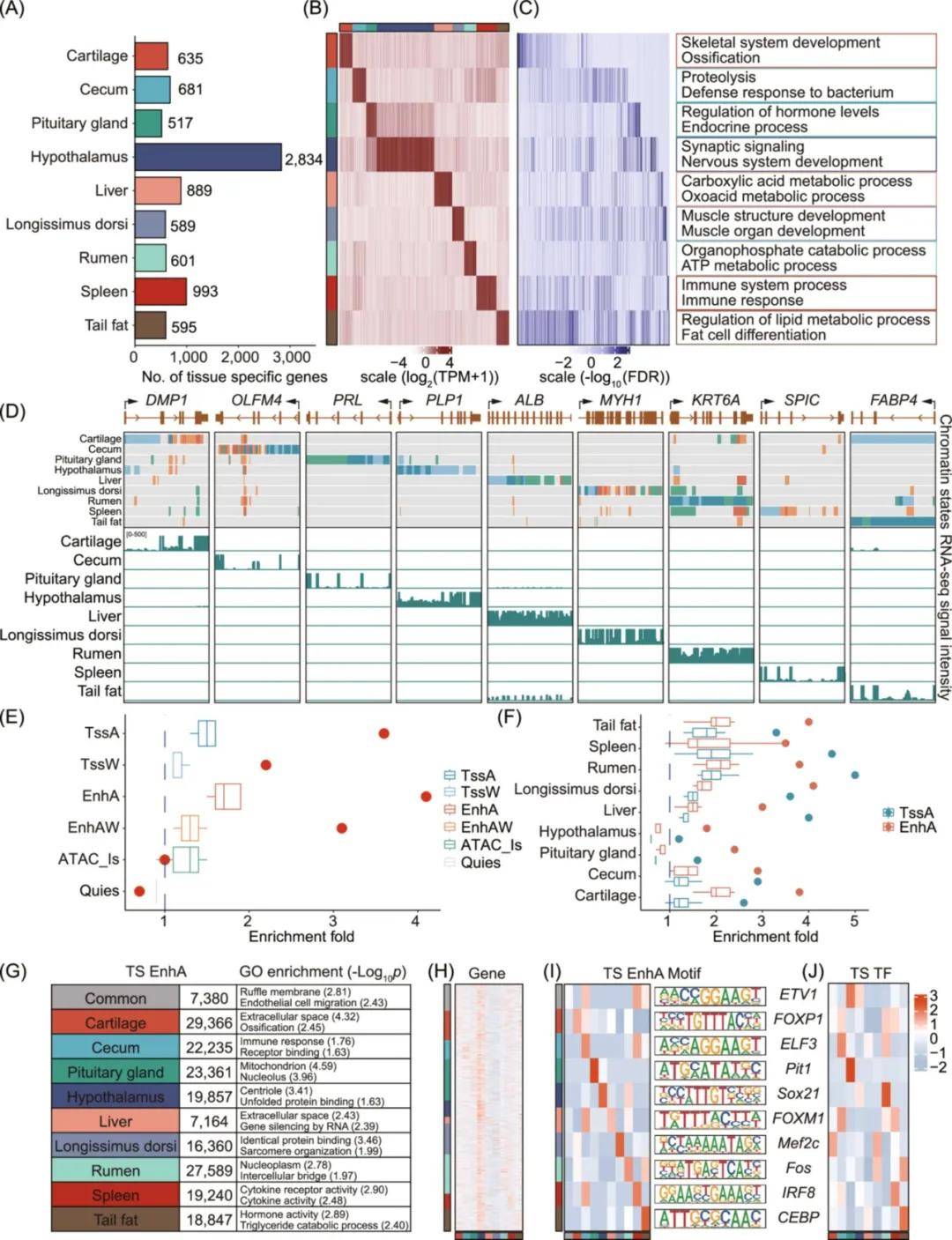 图3 组织特异性基因和染色质状态的功能表征
图3 组织特异性基因和染色质状态的功能表征
4、染色质状态预测,加深对绵羊驯化和改良过程中表型变异的理解
为探讨功能元件是否在受驯化和改良影响的基因组区域中显著富集。对来自全球的364只绵羊进行基于最大似然估计的群体结构分析。结果表明,所有个体被分为三个独立的簇:亚洲摩弗伦羊、中国地方绵羊品种和改良品种。剖析绵羊的驯化,计算了野羊和家养绵羊之间的固定指数(FST)、种群扩展单倍型纯合性(XP-EHH)和核苷酸多样性比率(π比率)。共确定了46个候选基因组区域,结果发现TssA、TssW和EnhA区域在受驯化影响的基因组区域中高度富集。在组织特异性染色质状态富集分析中,发现与免疫功能相关的脾脏特异性TssA和EnhA以及与感知能力相关下丘脑特异性TssA的富集程度较高。这一结果与绵羊在早期驯化过程中感知能力和生存能力被优先选择的事实相一致。
此外,对亚洲摩弗伦羊与中国地方绵羊品种(WN)、亚洲摩弗伦羊与改良品种(WI)和中国地方绵羊品种与改良品种(NI)进行FST、XP-EHH和π比率的比较分析。结果发现在WN、WI和NI比较中分别鉴别到49、53和13个候选基因组区域。富集分析结果显示,在NI组中盲肠、尾脂特异性TssA和背最长肌特异性EnhA高度富集,而脾脏、瘤胃和尾脂特异性EnhA在WN和WI组中高度富集。这与改良绵羊品种的育种目标相一致:更快的生长速度和减少尾脂沉积,而地方绵羊品种则是具有较强的适应性和抵抗力,这些结果表明功能元件在解释绵羊的适应性进化和复杂农艺性状方面发挥重要作用。
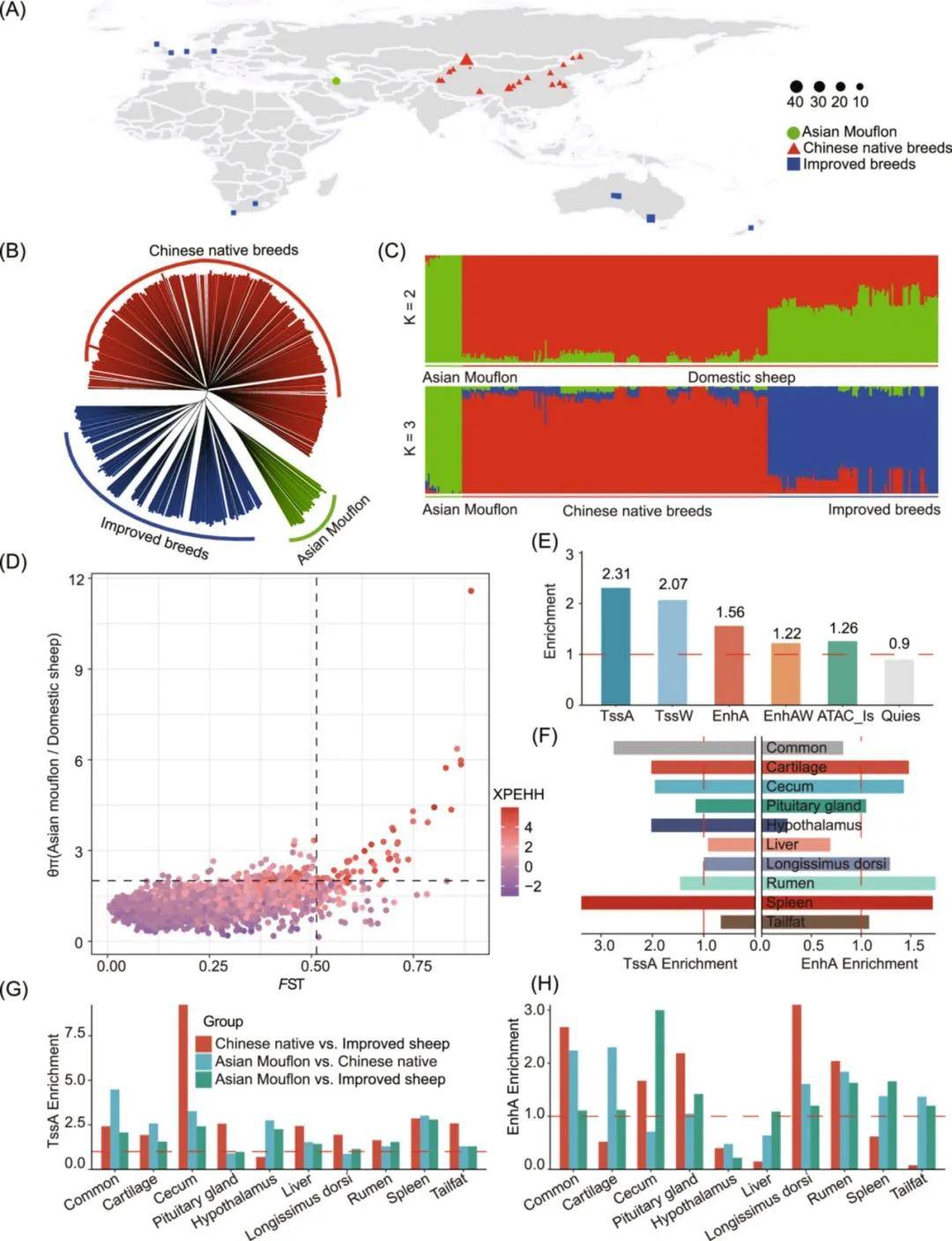 图4 绵羊驯化和改良过程中选定区域染色质状态的富集
图4 绵羊驯化和改良过程中选定区域染色质状态的富集
5、鉴定与绵羊尾部脂肪沉积相关的基因组区域
对168只脂尾型绵羊品种和170只瘦尾型绵羊品种进行了基于固定指数(FST)的选择信号分析,将前5‰的窗口定义为候选基因组区域。结果发现两个信号最强的区域分别位于Chr13(51.15-52.05Mb)和Chr15(3.67-4.50Mb)。进一步分别对长脂尾、短脂尾、脂臀尾、短瘦尾和长瘦尾绵羊品种两两进行选择信号分析,发现在不同脂尾绵羊和瘦尾绵羊种群的比较分析中鉴别到相同的选择区域。进一步对这两个基因组区域的等位基因频率进行统计,发现瘦尾和脂尾型绵羊群体中的等位基因频率分布存在很大的差异,表明这两个区域可能是影响绵羊尾脂沉积的候选基因组区域。
为缩小影响绵羊尾脂沉积的基因组区域,本研究对2,003只具有尾脂重量和尾脂相对重(尾脂重/胴体重)的大规模队列进行GWAS分析。根据-log10(0.05/总SNP)=8.63的阈值,在Chr7、Chr9和Chr13染色体上筛选出984个和1086个与尾脂重量和尾脂相对重显著相关的SNPs位点,其中981个SNPs与尾脂重量和尾脂相对重均显著相关,且位于Chr13染色体的受选择区域。值得注意的是,这些SNPs位于内含子和基因间区域,且处于强LD状态,因此有必要整合多组学数据来确定影响尾部脂肪沉积的因果变异。
 图5 不同尾型绵羊的全基因组选择信号分析
图5 不同尾型绵羊的全基因组选择信号分析
6、利用多维数据集对与尾部脂肪沉积的候选因果变异进行优先排序
为确定与尾脂沉积相关的候选因果变异并探讨其对尾脂沉积的作用,本研究整合了不同尾型的选择性扫描分析结果、尾脂重性状的GWAS结果和绵羊尾部脂肪组织的表观基因组数据(即ATAC-seq、H3K27ac、H3K4me3和Hi-C数据)。结果仅发现两个SNPs(Chr13:51760995A>C和Chr13:51825895G>A)位于OCR和H3K27ac峰。这两个SNPs也位于与骨形态发生蛋白2(BMP2)、LOC101117953和LOC101118027基因相同的拓扑关联域(TAD)中。结果表明这两个SNPs可能与绵羊尾脂沉积密切相关。
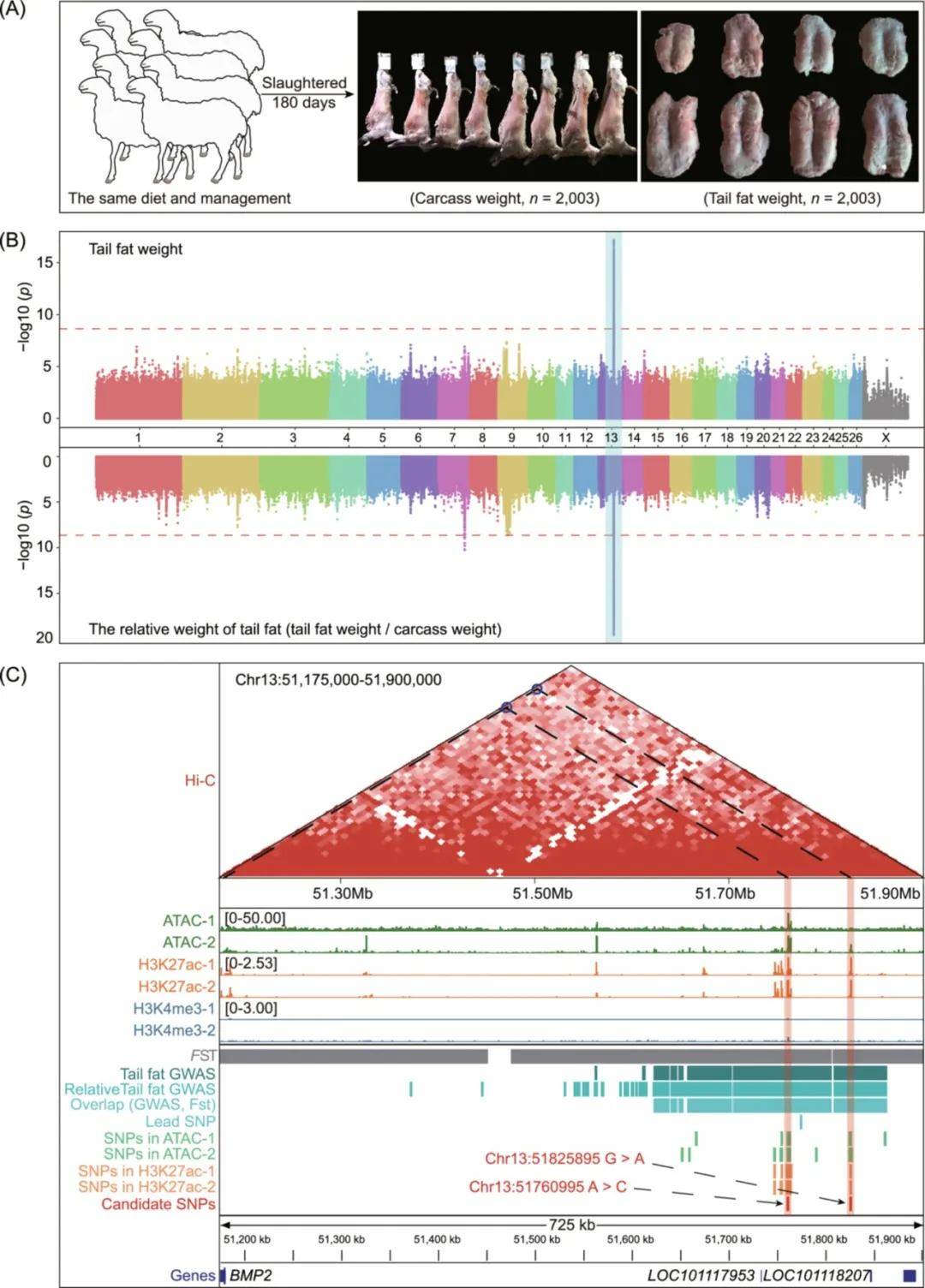 图6 整合全基因组关联研究(GWAS)和表观基因组数据,定位与绵羊尾脂沉积显著相关的候选因果变异
图6 整合全基因组关联研究(GWAS)和表观基因组数据,定位与绵羊尾脂沉积显著相关的候选因果变异
为进一步探讨候选变异如何影响绵羊尾部脂肪沉积,本研究统计了Chr13:51760995A>C和Chr13:51825895G>A SNPs位点在338只不同尾型绵羊群体中的等位基因频率。与脂尾型绵羊品种相比,Chr13:51760995A和Chr13:51825895G的参考等位基因在瘦尾型绵羊品种中的频率更高。携带参考等位基因(A/G)个体的尾脂重和尾脂相对重(尾脂重/胴体重)显著低于携带突变等位基因(C/A,p < 0.01)的个体。此外,对具有不同基因型绵羊的尾部脂肪组织进行转录分析,结果表明Chr13:51760995A>C的SNP位点的改变影响BMP2基因的表达。而Chr13:51825895G>A的突变不影响BMP2及其附近基因的表达。
为验证BMP2基因对绵羊尾脂肪沉积的影响,从尾部脂肪组织中分离原代前脂肪细胞,诱导其分化为成熟的白色脂肪细胞,构建过表达载体pcDNA3.1-BMP2,检测过表达效率、增殖及分化潜能。结果表明,BMP2过表达并不影响细胞增殖,但增强其成脂能力,表现为脂滴聚集和甘油三酯含量增加,以及脂肪细胞标志基因表达增加。相反,转染BMP2基因siRNA的细胞中脂滴聚集和甘油三酯含量与阴性对照组相比明显降低(p < 0.05)。综上所述,位于强激活增强子区域的Chr13:51760995A>C是一个影响绵羊尾脂重性状的可靠候选因果变异。
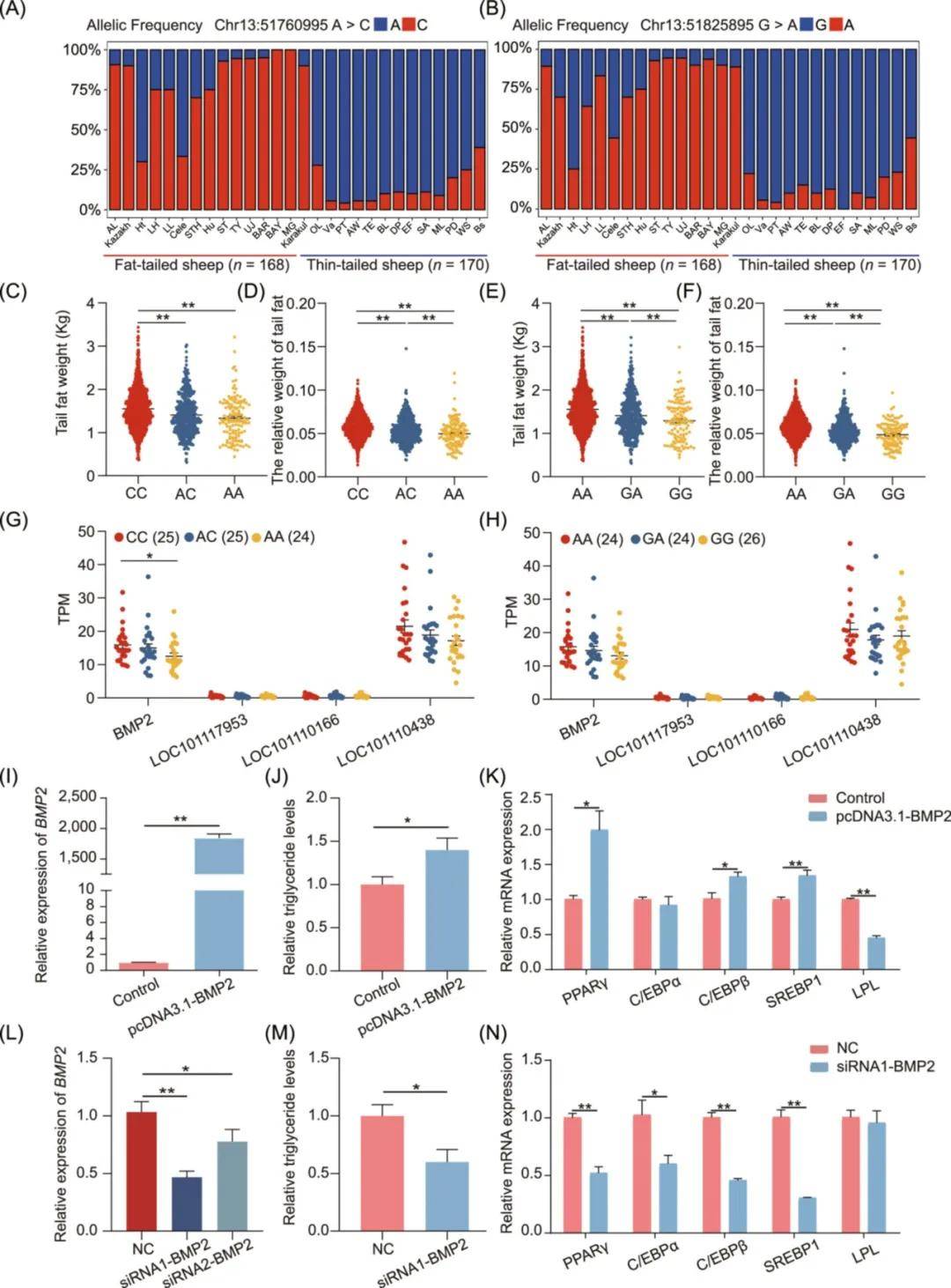 图7 候选功能位点鉴定及靶基因验证
图7 候选功能位点鉴定及靶基因验证
六、研究结论
本研究建立了首张绵羊多组织表观基因组图谱,填补了现有绵羊图谱数据集的空白。这为解析绵羊的适应性进化和复杂经济性状提供了基础数据资源。同时,通过对多组学数据集的整合分析,鉴别了一个新的因果突变(Chr13:51760995A>C),该位点可作为绵羊育种计划中减少尾脂沉积的潜在遗传标记。总之,这些发现为绵羊育种提供了宝贵的资源和分子标记,有助于加速遗传改良。
原文索引
https://doi.org/10.1002/imt2.254


















