

2017-03-08
以PacBio公司的SMRT单分子实时测序技术(Single molecule real-time sequencing)为代表的三代测序技术,通过其独有的环形一致性测序模式(Circular-consensus sequence,CCS),极大提高了单碱基测序的准确率,远超Illumina等二代测序技术。与传统转录组测序项目相比,利用PacBio平台的全长转录组测序技术可以直接获得mRNA的全长,保证了mRNA序列的精确性。上期我们为大家介绍了全长转录组测序的数据质控,本期将为大家介绍三代全长有参转录组的分析内容。主要包括:
1. 转录本分类
数据过滤后得到高质量的Reads of Insert(RoI),接着对其进行转录本分类。理论上完整的RoI应该有5’primer、3’primer和polyA部分。但在实际测序中,由于5’端降解等因素,并不是所有的RoI都是完整的,即不是全长转录本序列,而且有少部分RoI序列为嵌合体。因此通过检测RoI序列是否含有 5’primer, 3’primer和polyA以及其位置的关系, 将RoI序列分为全长非嵌合(Full-Length-Non-Chimeric)序列、全长嵌合(Full-Length-Chimeric)序列、非全长(Non-Full-Length)序列等。其中全长非嵌合序列即是物种原始的全长mRNA序列,各分类之间的相互关系见下图:
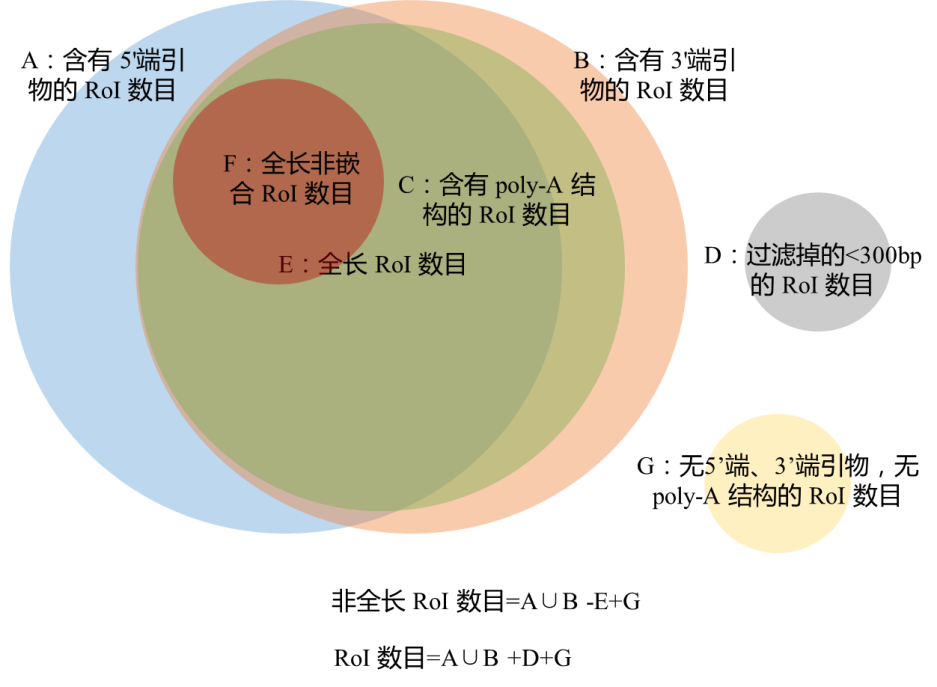
图1 转录本分类
2. 序列聚类与矫正
全长非嵌合序列中存在大量的冗余序列,我们将冗余序列聚类到一起,得到新的一致性序列(Consensus Isoforms),然后将非全长序列比对到一致性序列上进行校正,最终得到准确度大于99% 的高质量Isoform(即转录本)。下图是Isoform长度分布图,长度分布的区间与文库片段的选择有关。
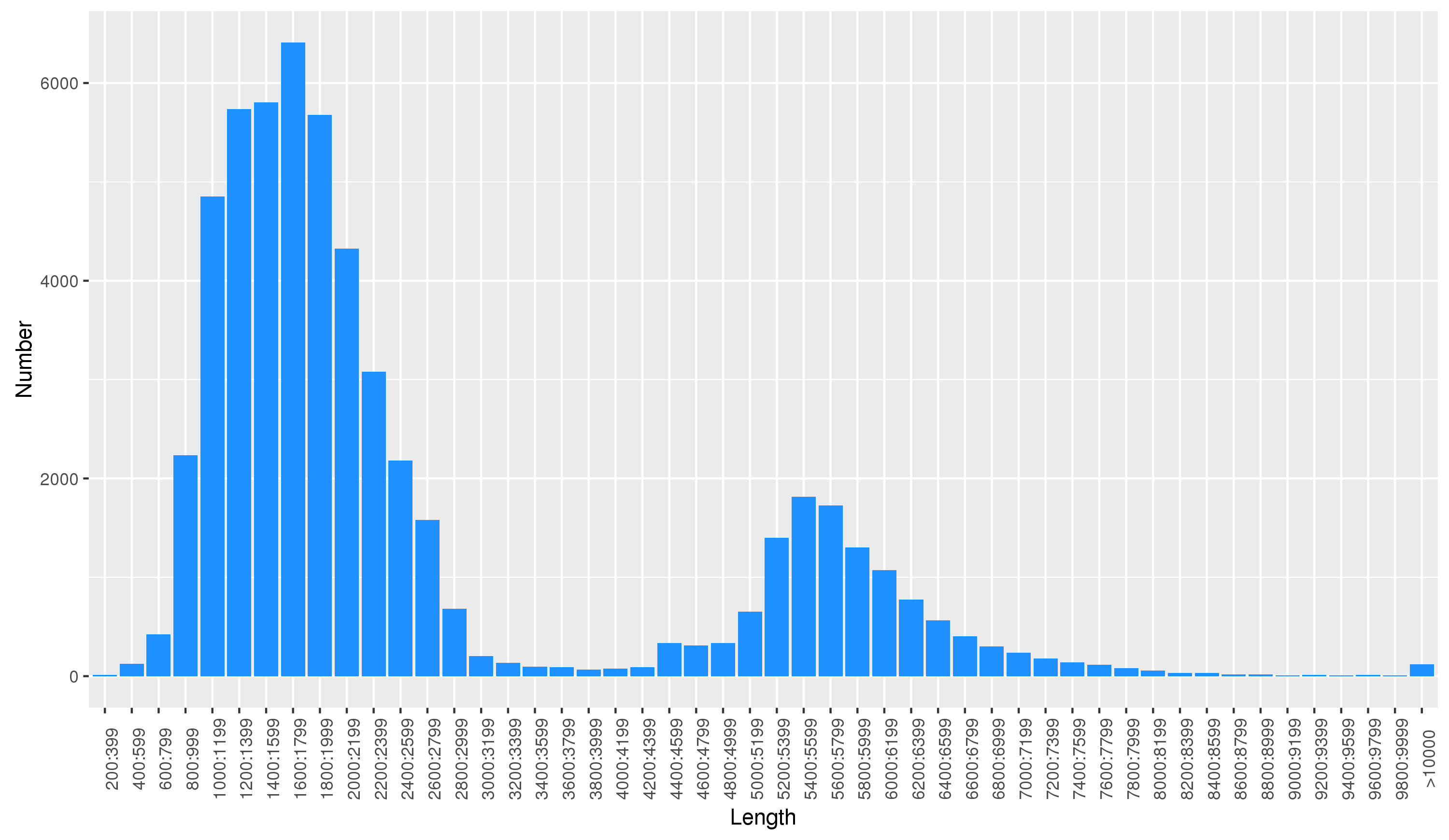
图2 Isoform长度分布
3. 参考基因组比对
我们将经过校正得到的Isoform与参考基因组比对, 根据基因组注释信息,可得知序列的来源基因以及表达产物的结构。与参考基因组比对情况见下图,图中Score 0:覆盖了基因的区域,但是没有或者少有与基因的exon匹配上的Isoform数目;Score 1:与基因的一些exon有一对一overlap的Isoform数目; Score 2:与基因的一些exon有一对一overlap并且匹配情况较好的Isoform数目。
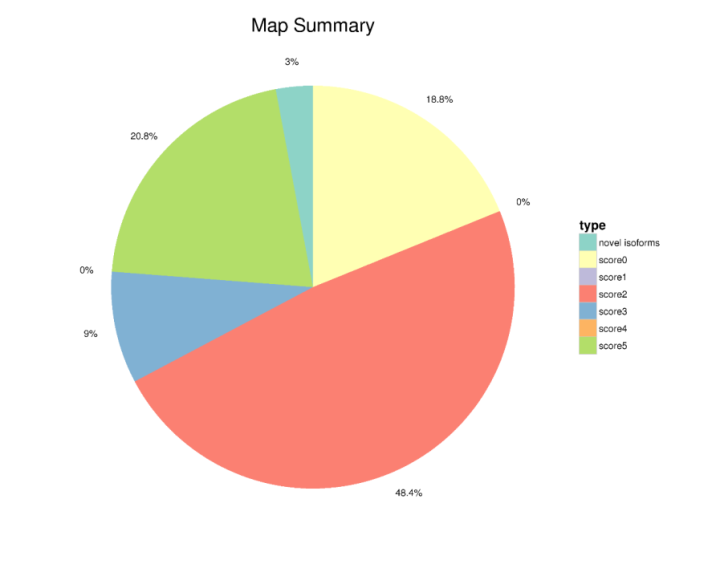
图3 比对参考基因组情况统计
4. Known Isoforms 和Novel Isoforms功能注释
对于比对到基因组上的score为0-5的known Isoform采用有参考基因组注释方法,对于Novel Isoform采用无参考基因组注释方法。基因功能注释所用到的数据库包括 GO、KEGG、eggNOG、NR、Swiss-Prot等。下图是其中的eggNOG注释结果:
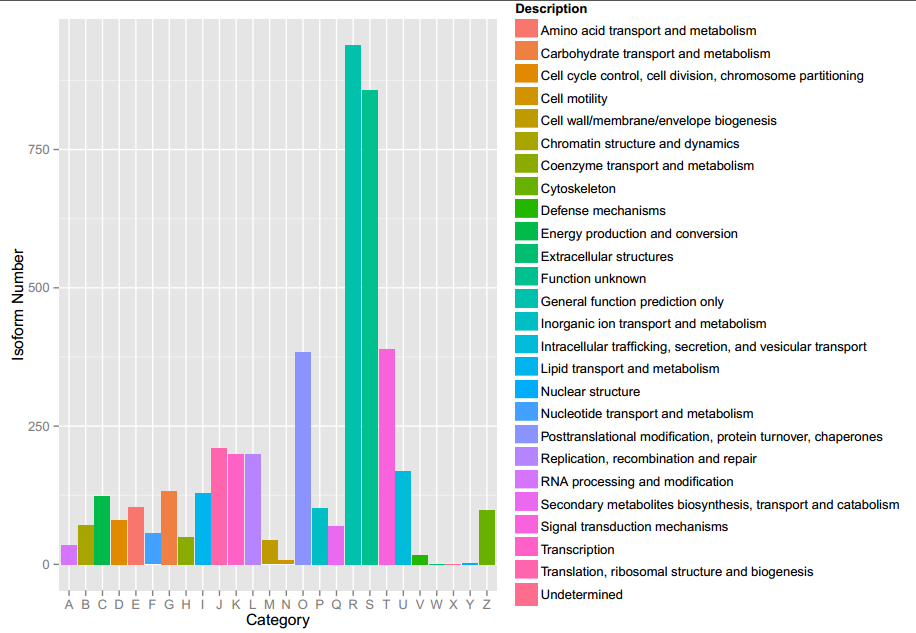
图4 eggNOG注释
5. 结构分析
结构分析是三代全长转录组中的一个重点研究内容,不同的样本转录物不尽相同,我们进行转录本结构分析,可以统计所有转录本的结构差异。它能够准确辨别二代测序无法识别的同源异构体或同源基因等。转录本结构分析包括可变剪接,融合基因, UTR区域注释,cSNP 和InDel 分析等。
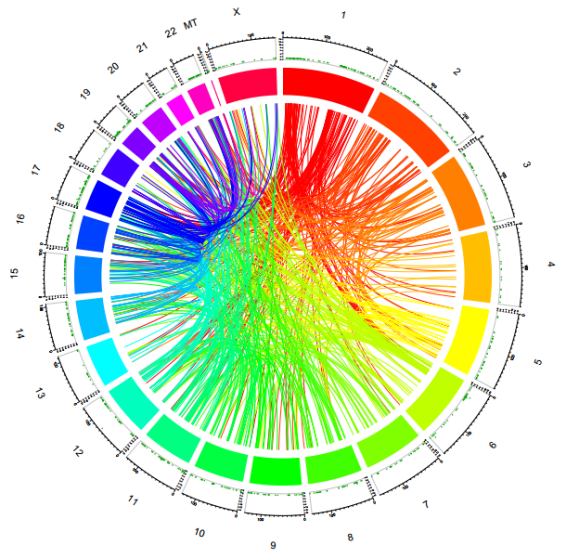
图5 融合基因分析
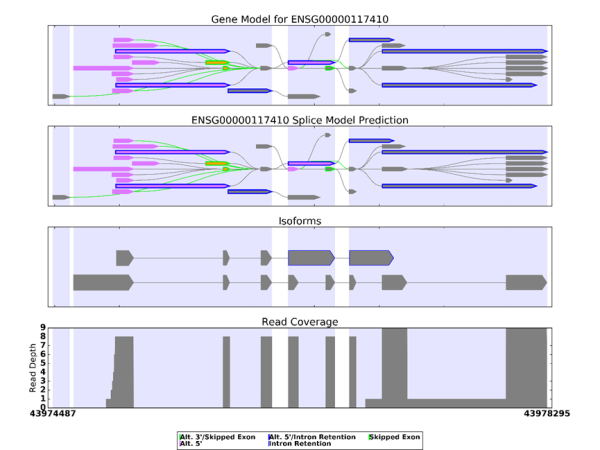
图6 可变剪切分析


















