

2017-03-01

该研究于2016年发表于Nature子刊《Scientific Reports》上
(最新影响因子:5.228)
研究背景:
拼接组装一直以来都是宏基因组项目中最重要的一个环节,它对后续的基因功能注释、微生物基因组重建以及物种分类注释都起到很大的影响。目前,Illumina平台较短的测序片段对宏基因组拼接结果影响较大,而三代基于PacBio的SMRT单分子实时测序技术的兴起,将有望显著提升宏基因拼接组装的效果。
研究方法:
样本来源:沼气反应池微生物群落
测序平台:PacBio RS II+Illumina HiSeq
通过Hiseq和PacBio平台的测序分析,比较两个平台的宏基因组组装的效果以及物种注释精确度。
研究结果:
Hiseq平台产出18.5 Gb宏基因组数据用于拼接,共拼接得到3,035,577个Contigs,平均189 nt,55,633个Contigs> 1 kb,最大长度148,797 nt。而利用PacBio平台产出的95.4 Mb数据用于拼接,共拼接出2,181 个contigs,平均长度4,459 nt,最大长度65,165 nt。
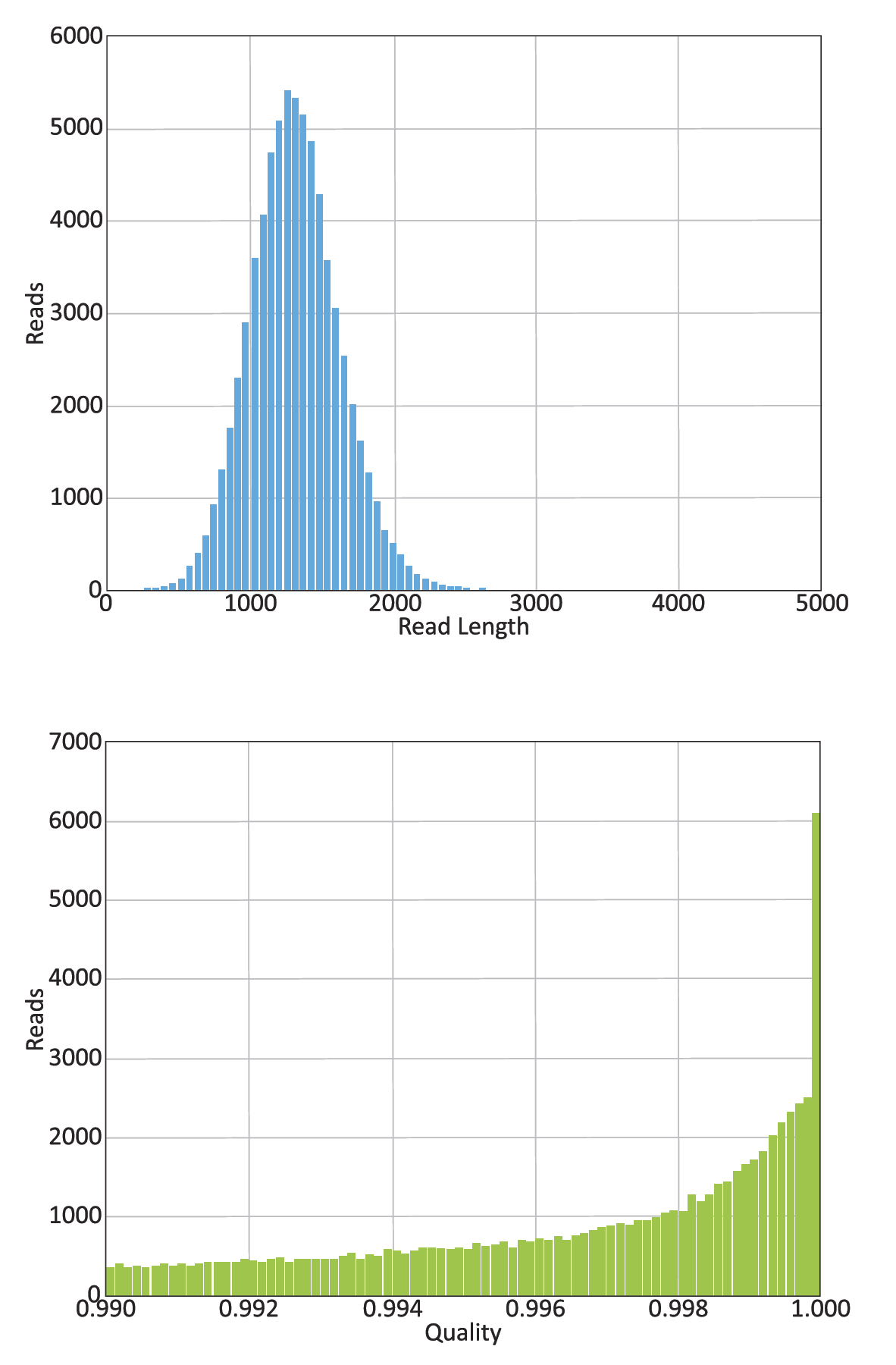
采用PacBio RS II测序平台中的P4-C2试剂盒,插入1.5kb片段构建文库进行CCS测序,在获得的测序结果中,上图为质量分数高于99%的序列长度分布统计以及序列质量分布统计,其中共71,254条序列质量高于99%,平均序列质量高达99.7%。
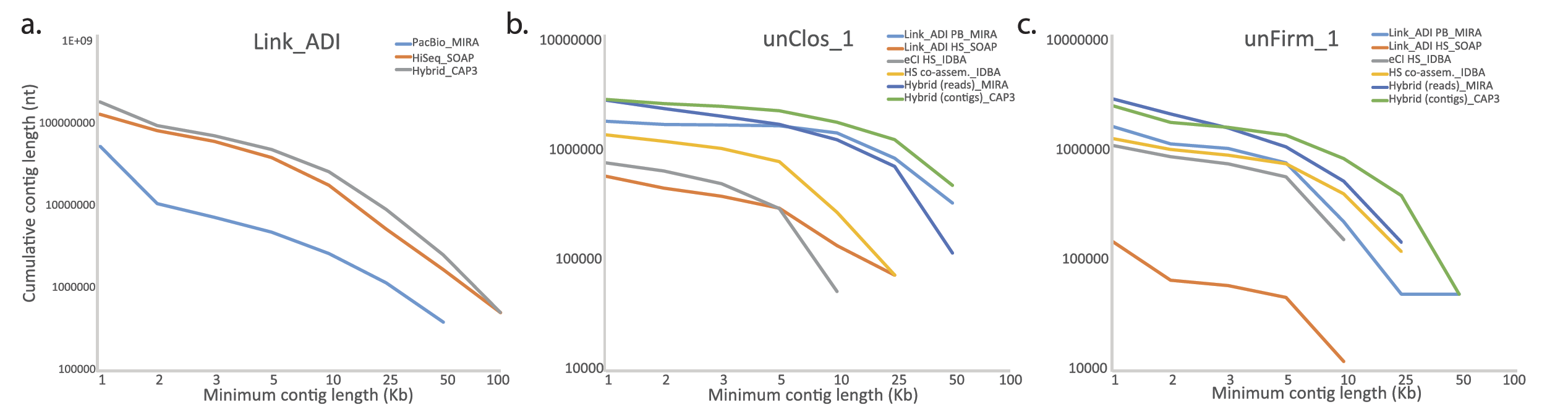
不同测序平台、不同拼接软件对宏基因组测序数据组装拼接获得的Contig长度比较。
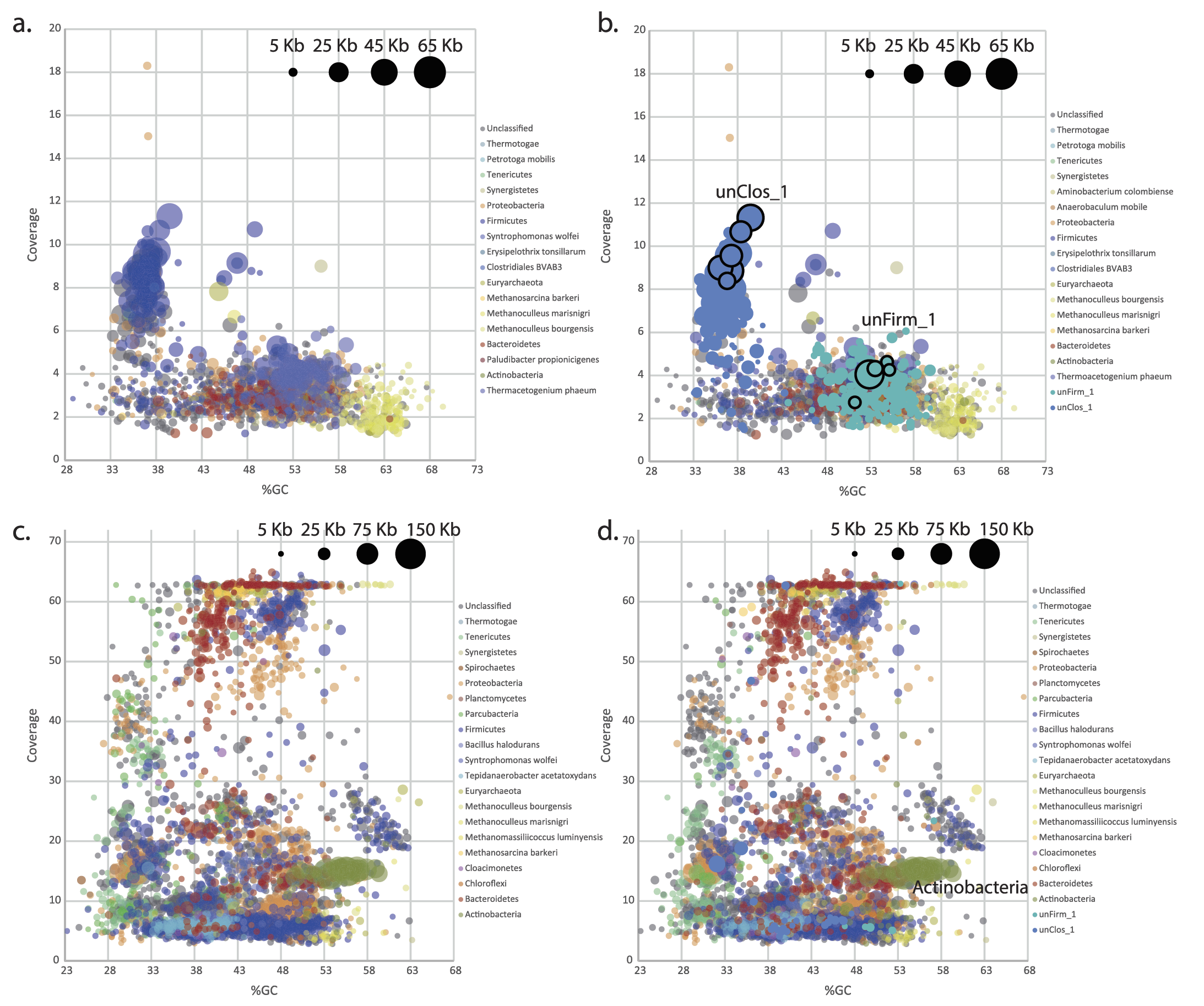
对同一份样本分别采用PacBio(a-b)和HiSeq(c-d)测序,运用PhyloPythiaS进行物种分类注释,并对注释结果(GC含量、覆盖度和Contig长度)进行可视化比较。由图可见,三代PacBio平台获得的Contigs更长更完整,二代HiSeq平台获得的Contig碎片多得多。b图和d图在注释分析时加入了物种特异性的训练数据集。
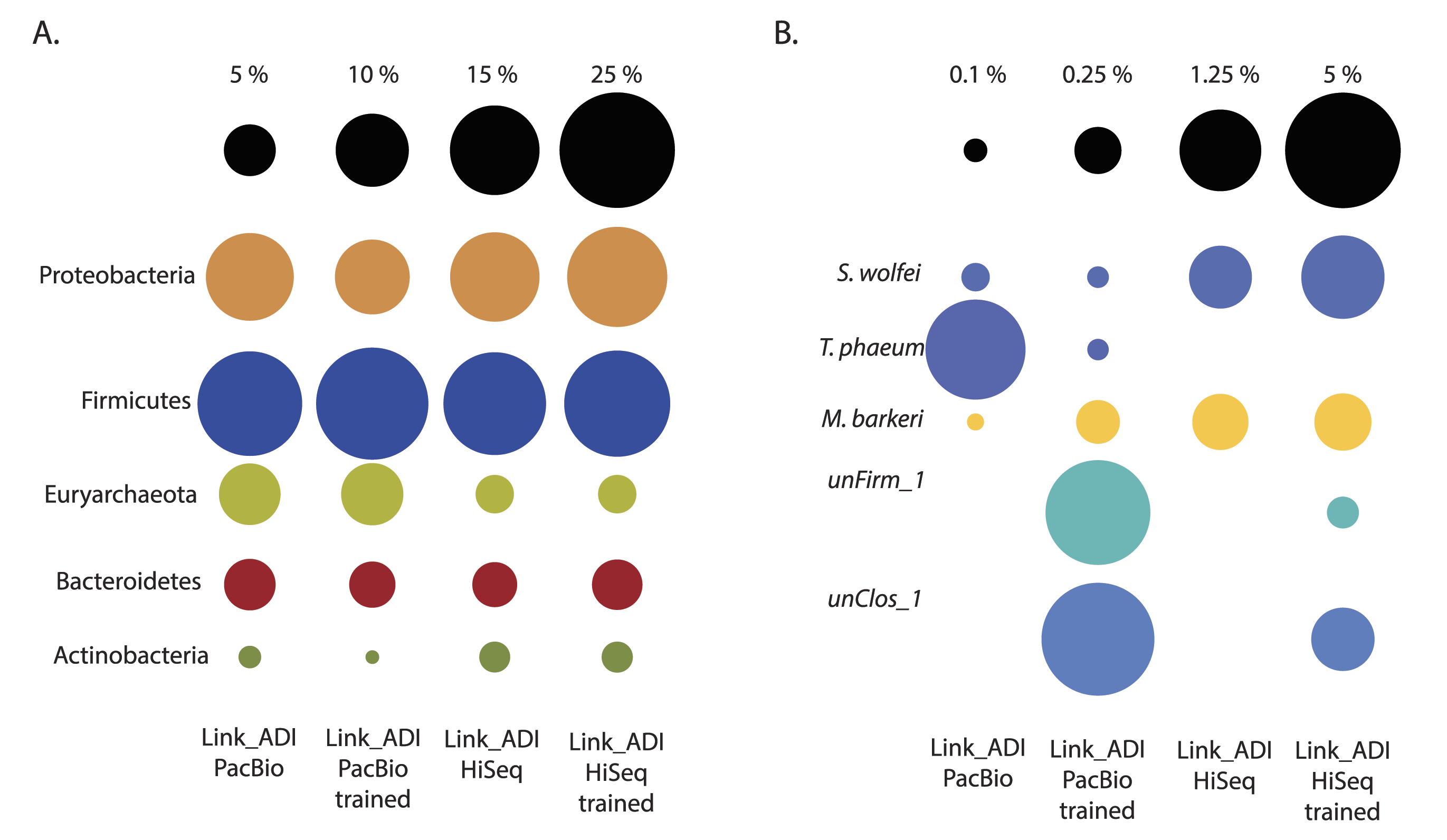
对两种不同平台物种分类注释结果的比较,分别基于(A)图门水平与(B)图种水平的物种丰度比较结果。由图可知,在门水平,两个平台测序数据的物种丰度较为一致,但在种水平,不同物种的丰度差异比较明显,尤其在加入了物种特异性的训练数据集后,PacBio平台对于unFirm_1与unClos_1两个物种的丰度结果统计更加精准。
研究结论:
综上所述,运用三代PacBio测序技术,将显著提升宏基因组数据的拼接效率和物种注释精准度,相比二代Illumina HiSeq测序平台,可谓是取得了质的飞跃!
派森诺优势
2016年,派森诺生物在原有的PacBio RS II三代高通量测序仪基础上,率先部署最新款Sequel测序仪,并已投入使用,独家提供三代测序分析服务,助力微生物组研究!
作为行业先锋,派森诺生物将一如既往地行使“解析序列,诠释生命”的理念,秉承“立足客户需要,满足个性需求”的服务宗旨,始终如一地提供性价比最高、最优质、最快速稳定的高通量测序和数据解析方案。
派森诺生物将竭诚为您服务!
参考文献
Frank, J. A. et al. Improved metagenome assemblies and taxonomic binning using longread
circular consensus sequence data. Sci. Rep. 6, 25373; doi: 10.1038/srep25373 (2016).


















