

2016-09-08
通过之前小编对三代全长转录组测序的专题介绍(终于等到你......三代全长准路阻测序技术原理篇/三代测序将为转录组研究者带来哪些惊喜),各位“诺米”们应该也了解到,利用三代基因测序平台PacBio RSII/Sequel系统能获得mRNA全长,更为精准地解读mRNA的结构信息。可实现同源异构体分析,单碱基变异检测,可变剪接分析,检测融合基因,等位基因识别等。
可能“诺米”们会质疑,为何我们单页里要专门强调分段建库呢?那是因为基于PacBio平台的测序原理,构建好的全长文库要loading到测序小孔——零模波导孔(ZMWs),由于mRNA长度不同,在loading的过程中会产生loading bias,即测序小孔会优先被长度较短的片段占据,每个测序小孔只能容纳一个文库分子,而大部分长片段则会“无家可归”。因此为尽量降低loading bias的影响,需要根据测序物种mRNA的长度进行分段,使一个文库中的序列长度控制在一个较窄的范围内。
通过小编的讲解,“诺米”们是否茅塞顿开呢!而关于文库方案的选择,小编根据已有的一些文献做了整理以供参考:
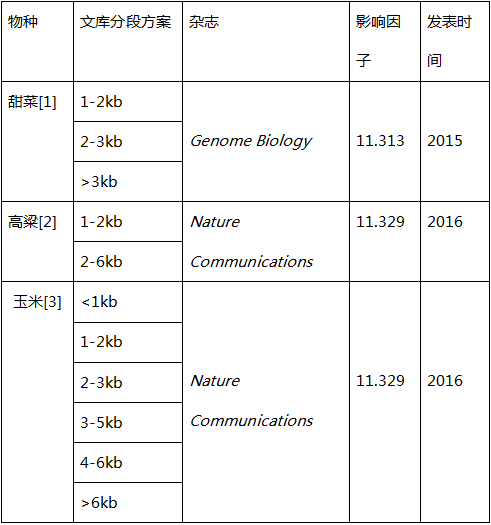
可见对于文库的分段方式,并不是固定的。
文库的数量和分段的形式可根据实际情况进行选择和优化,主要考虑如下因素:
物种转录本的长度分布;
所关注的mRNA长度范围;
mRNA的覆盖度需求。
如需更好的实现mRNA的均匀覆盖,在保证测序数据量的同时,最好尽可能多的构建文库。而关于数据量的要求,将在下期跟各位“诺米”讨论,敬请期待!
参考文献:
1. Minoche A et al., Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biology (2015) 16:184
2. Abdel-Ghany S et al., A survey of the sorghum transcriptome using single-molecule long reads. Nature Communications (2016) 7:11706
3. Wang B et al., Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nature Communications (2016) 7:11708


















