1.DNA测序的原理是什么?步骤如何?ABI 3730XL测序的准确程度如何?
DNA测序的原理是末端终止法。具体步骤如下: 1. 定量:对样品及引物进行定量。 2. 测序反应:在反应体系中加入已定量的模板和引物,BigDye等试剂,PCR仪上完成测序反应。 3. 读取数据:根据荧光的不同,读取被测样品的碱基序列。 ABI公司目前测序结果在800bp以内准确率为98.5%。
2.为什么在测序报告上找不到我的引物序列?
如果您提供的样品是PCR产物,在测序报告上找不到您的引物是正常的。这是因为测序反应是通过对被荧光标记的ddNTP的读取而获得基因序列的,而测序引物由于没有荧光标记,因此自然在测序结果中就不会被识别。有两种方法可以得到您的引物序列。对于较短的PCR产物(<800bp),可以用另一端的引物进行测序,从另一端测序可以一直测到序列的末端,就可以在序列的末端得到您的引物的反向互补序列。对于较长的序列,一个测序反应测不到头,因此就只能将您的PCR产物片段克隆到适当的载体中,用载体上的通用引物进行测序。由于载体上的通用引物与您的插入序列之间还有一段距离,因此就可以得到您的完整的引物序列。由于在测序的起始端总会有一些碱基无法准确读出,因此,您如果想得到您的PCR产物的完整序列,克隆后进行测序。 如果您提供的样品是质粒或菌液,PCR产物用T载体克隆后,由于克隆的方向是随机的,因此,当您在一条链上找不到您的引物序列时,试图在互补链上寻找您的引物序列。 当测序引物离您的插入片段很近时,有时可能也无法找到您的引物的全序列。这主要是因为有时测序的起始端由于未去除的染料或引物二聚体的干扰,造成起始区的序列不好,可能无法找到您的引物完整序列。 有时,质粒做模板进行测序时,由于某些原因,质粒上没有插入外源片段,为空载体,所测的序列为载体序列,此时自然也找不到引物序列。
4.过短的PCR产物为什么不适于直接测序?
首先过短的PCR产物纯化困难,一般的PCR产物纯化试剂盒都要求PCR产物片段大于200bp,过短的PCR产物纯化和准确定量都非常困难。因此我们要求用于测序的PCR产物一般不低于200bp长度。 其次,由于测序本身的限制,以一个100bp的PCR产物用于测序为例,去掉两个引物的序列大约40到50bp,再加上测序起始端的一些读不出的碱基,真正能够得到的有用序列不过30~40碱基。上面是在理想的条件下的假设,稍不顺利,测序就失败了。因此,过短的PCR产物,只能克隆后进行测序。
5. 我的引物做PCR效果很好,为什么用于测序总得不到好的结果?
用于测序的引物比用作PCR反应的引物要求高,以下是不适合用于测序反应的引物类型: 1. 不纯的引物:用于测序的引物纯度要求很高,合成中的小片段会直接造成严重的背景峰,因此用于测序的引物序列长度一般在24个碱基之内。 2. 简并引物,随机引物和有特殊标记的引物。
6. 我进行PCR反应时,所用的退火温度是59℃,是否可以用该温度对我的样品完成测序反应?
非常抱歉,目前我公司不会单独对客户的测序反应设定特定的条件,测序反应均在统一的条件下完成。
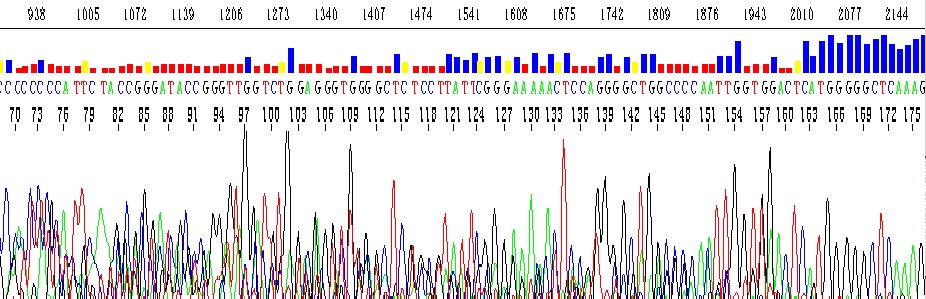
9. 我的PCR产物电泳检测条带单一,为什么测序结果说模板杂?
PCR产物电泳检测的结果只是一个粗略的定性结果。对于与目的片段条带大小只相差几个碱基的非特异性PCR扩增产物是无法用肉眼区分开的。但是DNA测序反应敏感而客观,可以直接反应出模板本身的情况。需要强调的是,高质量的PCR产物才可能得到高质量的测序结果,如果您对PCR产物的纯度不很确定,希望您可以将PCR产物进行克隆处理,得到好的测序结果。以下图为例:该反应的背景信号较高,不利于碱基的判读。
解决办法:改变PCR条件,重新扩增。或者可以将该PCR产物克隆到质粒中,初步筛选后进行单克隆测序。
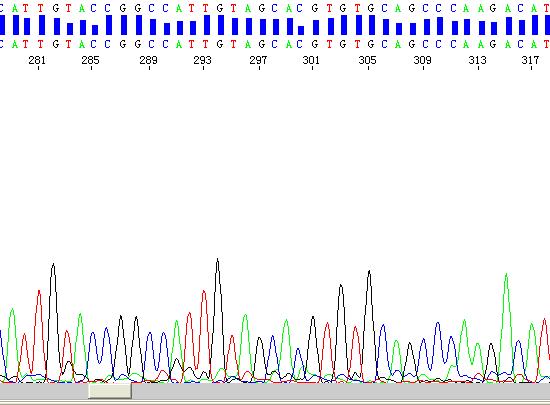
10.什么是碱基缺失?
以下图为例:
碱基缺失常见在PCR产物中,特别是从基因组中扩增得到的PCR片段,上图的186位缺失两个连续的T。
解决方法:1) 使用反向引物继续测序,以矫正缺失位点并达到测通的目的。或者将该PCR产物克隆到质粒中,挑取单克隆测序。
2) 如果可以确定该PCR片段中不应该有缺失的位点,那么可以改变PCR反应条件,重新扩增。
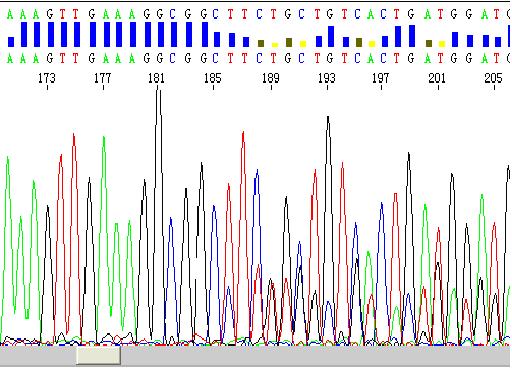
11.什么是引物不纯?
以下图为例:
引物不纯造成移码现象,该种现象与模板杂在峰图都表现为背景峰较杂,但是引物不纯在峰图上表现的更有规律,一般在每一个主峰前都有一个同一碱基的小峰。
解决方法:重新合成引物,或者将引物进行PAGE纯化后再进行测序。
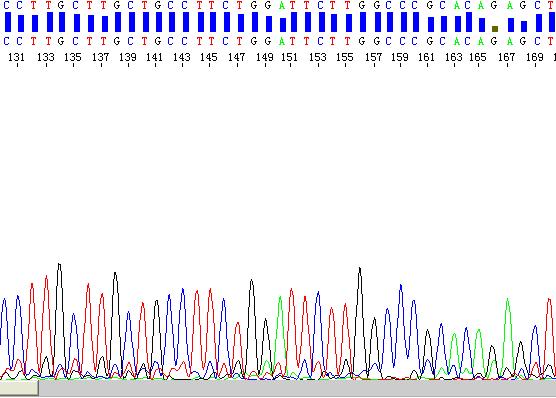
12.重复结构对测序有哪些影响?
以下图为例:
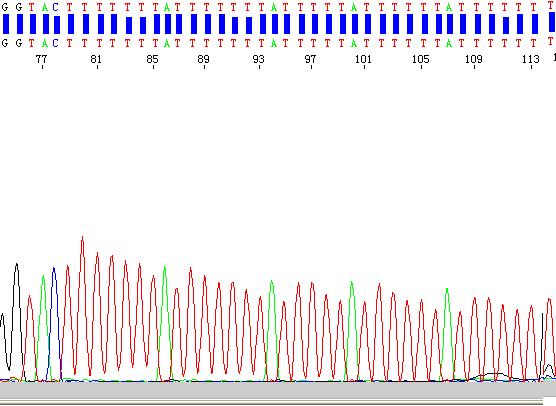
重复结构将导致测序复制框的滑移,重复结构之后峰型混乱。
解决办法:使用反向引物对模板进行测序,测到该重复结构处,即可完成模板全长的拼接。
13.什么是模板不单一?
以下图为例:
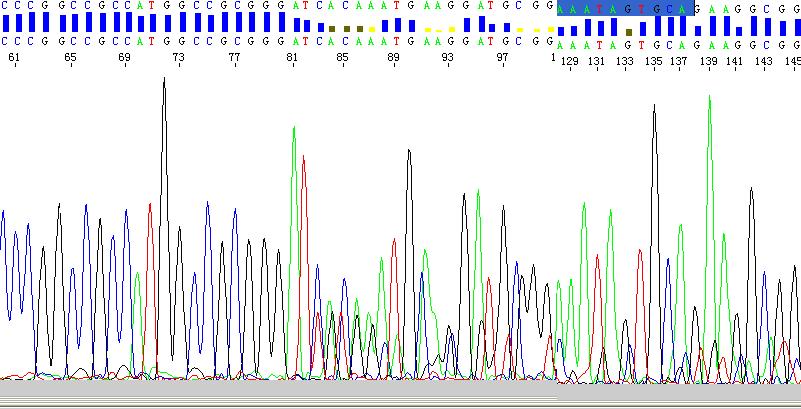
上图是pGEM-T载体测序的结果,在83位点处测序结果出现双峰,即测序结果在载体部分很准确,而进入插入片段后出现双峰的情况。这是由于在接种时没有挑单菌落导致的,当两个以上的正常的克隆(插入片段方向相反),或正常克隆与空载体混在一起,而通过酶切和PCR鉴定很难看出异常,尤其在T-A克隆时经常碰到。
解决办法:重新涂平板挑单菌落测序。需要注意的是,重新进行PCR反应或者酶切鉴定仅能证明该克隆含有插入片段,并不足以证明模板的单一。
对于PCR产物也同样有模板不单一的情况,如下图所示,
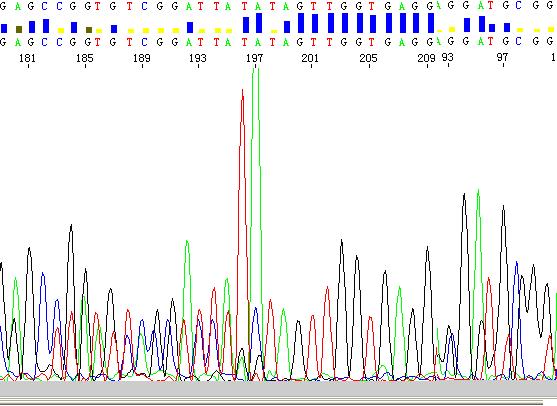
在197bp前测序峰表现为杂或有明显套峰,且在197bp位置有一个高高的A峰,峰标志着此PCR产物中有一个片段大小为200bp左右的小片段。
解决方法:对PCR产物切胶纯化,再进行测序。
14. poly结构的测序结果是怎样的?
以下图为例:
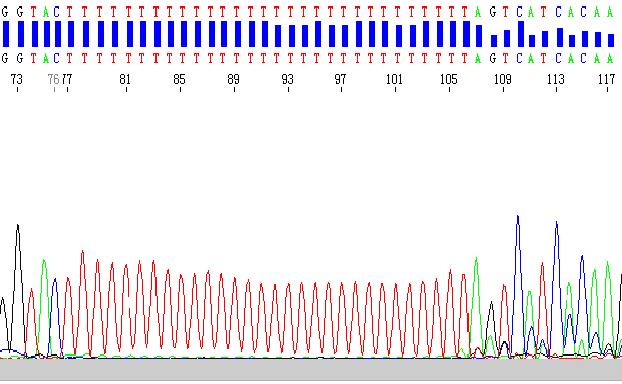
以polyT为例,在polyA/T结构之后往往出现移码现象,而在polyG/C之后会往往导致测序信号的衰减。
解决办法:使用反向引物对模板进行测序,测到该poly结构处,即可完成模板全长的拼接。
15.含有回文结构的模板测序结果是怎样的?
以下图为例:
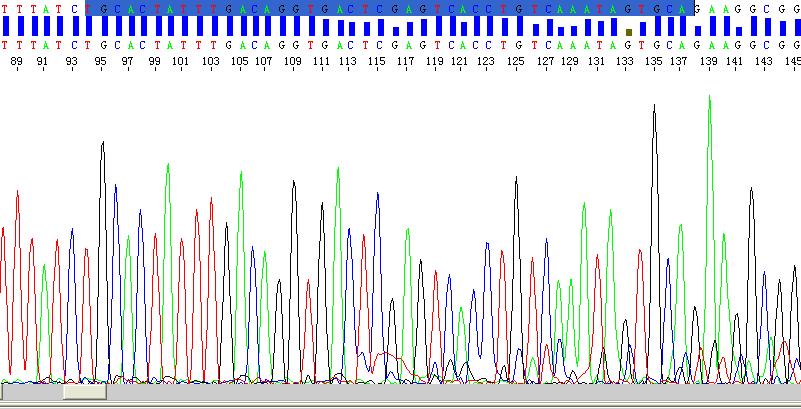
位点94至137是一个回文结构,该结构导致后面的信号衰减,出现错误的判读。
解决方法:使用反向引物对模板进行测序,测到该回文结构处,即可完成模板全长的拼接。
16.盐分高的情况是怎样造成的?
盐分高是指客户提供的PCR产物中的盐离子浓度高。造成样品盐离子高的原因是由于客户在进行PCR反应时,在反应体系中加入的盐离子浓度过高。盐离子的来源有PCR反应体系中的缓冲液即buffer,有些情况下是由单独加入的镁离子造成的。对于这种样品,通过电泳检测是不能对其进行区分的,只有在测序结果出来后分析峰图才可能发现问题所在,典型的峰图表现为前70-80甚至100个碱基的峰型基线漂离,严重影响测序结果前面部分的准确性。由于通过纯化的方法只能部分的去除少量盐分,无法解决盐离子对测序结果的影响。